The DAS website is at: http://biodas.org/ and contains the full DAS specification.
One goal of any genome project is the elucidation of the primary sequence of DNA contained within a given species. While the availability of the primary sequence itself is valuable, it does not reach its full potential until it has been annotated. Generally defined, annotation is descriptive information or commentary added to text, in this case genomic sequence. As the sequencing projects proceed and finish, the focus shifts toward annotation [30].
Full genome-scale annotation is a difficult problem. Some technical challenges include the sheer volume of data, the heterogeneous and growing types of annotations, the time sensitivity of searches, and the need to present the information in an integrated graphical fashion. A wealth of information is contained in the laboratories and individuals within a research community. Each laboratory wants to use its field of expertise to record insights about a portion of the primary sequence. Without a mechanism for collecting, recording, and disseminating this community-based annotation, a valuable source of information is severely diminished [39].
We propose to design and develop a distributed annotation system (DAS), allowing interested laboratories to develop and maintain annotations which are readily accessible to the community at large. The system needs to be easy to use, readily accessible by the community, and capable of representing annotations graphically.
This solution has proven to be unsatisfactory. Even in the best automated environments, high quality data sources require some human intervention in the annotation process. Experimental results are a critical component of annotation, an aspect overlooked by the Wheeland and Boguski definition. In addition, researchers ``want communication at least as much as they want information access'' [33]. This communication is informal and often takes the form of an annotation. To maintain quality, data sources need to be heavily curated to reflect new developments, additional knowledge, and continuing research efforts.
Currently most databases encourage users to submit annotations and changes to the data centers for inclusion. A curation group then reviews the information and decides what and how it is to be incorporated. The centralized nature of this system limits community involvement. Consequently, even in tight-knit communities, many useful annotations maintained in individual's laboratories never get incorporated into the official release. Thus far, attempts to improve community involvement have centered around consolidating current database systems. Classical approaches to managing heterogeneous databases include connecting them using the world wide web, organizing them into database federations and data warehouses, or dumping their data into centralized data repositories [37,27].
Many of the biologically relevant databases provide WWW connectiveness. For example, Proteome Inc.'s WormPD [24] provides links to AceBrowser, GenBank, the Protein Information Resource (PIR) [5], and Swiss-Prot [4] when available. Links are a one way navigation tool and require high maintenance. To utilize the information contained in these cross-linked systems requires familiarity with multiple database systems. These limitations create a barrier to efficient information dissemination and usage [27].
Database federations and data warehouses are similar methods which entail developing a global schema, the basic underlying structure, of the component databases. The principle difference between a federation and a warehouse is the way they integrate data. A federation uses an approach known as lazy integration, where data are retrieved, processed, and returned after each query. In contrast, a warehouse uses an eager approach, retrieving and integrating data in advance of a query. The tradeoff between database federations and warehousing is one of query performance versus data ``freshness''. The need for a strict common schema has led to social and technological difficulties in building and maintaining these systems [58]. One system in molecular biology is the Sequence Retrieval System (SRS) [18]. SRS deals with semistructured ASCII text by parsing and indexing mechanisms which require a controlled vocabulary (schema). The indices are warehoused but the data is federated, creating a hybrid system.
Another method of data integration is through data repositories, monolithic databases open to insertion and/or modification by the community at large [1,9]. In principle, making a database available to community annotation is an open and democratic method of removing the curation bottleneck. In practice, it raises tremendous problems with annotation quality, accountability, data ownership, and data integrity [17]. One of the earliest repository systems was the Worm Community System v2.0 (WCS) [49]. This system allowed community annotations to be written to a central database. However, the WCS failed to gain wide acceptance in the community due to competition from a competing database system, ACeDB, and the emerging WWW, among other factors.
Over time, steps have been taken to overcome many of ACeDB's initial limitations. In 1995, a text-ACE (TACE) [40] was introduced to allow shell scripting interactions. In the years following, WebAce [56], and Ace.pm (Steve Rozen, unpublished) provided richer scripting interaction with the ACE data. More recently, Lincoln Stein has developed two complementary libraries, JADE [51] and AcePerl [52]. They utilize the power of the new ACE servers to provide access directly to ACE objects from Java and Perl, respectively. The database-specific details are hidden by these libraries, providing a much needed level of abstraction. AceBrowser [53], a set of common gateway interface (CGI) scripts, was developed by Lincoln Stein from AcePerl. It provides some of ACeDB's functionality though the WWW.
Ideally, sequence annotations must be presented graphically and in an interactive display. This allows the user to adjust the granularity of information presented and to explore by requesting more details on regions of particular interest. A variety of annotation visualization tools already exist including Entrez [19], Chromoscope [6], Genome Topographer [15], and Anubis [41]. These tools differ in their portability, file formats, map presentation, and ease of use. At the other end of the spectrum are independent display components. The bioWidgets Consortium [12] and Neomorphic's [23] Java package known as the Genome Software Development Kit (GSDK) are just two examples. These tools simplify programming of visualization components.
One group particularly interested in annotation is the digital libraries community. Studies of annotation [38,39] demonstrate that in general annotation has value and users are typically aware of what kinds of material they trust as annotation. Libraries exist to serve the research needs of their constituents. Consequently, digital libraries aim to foster informal collaborations and communication through fluid and transient materials, including annotations.
The collective experience with traditional libraries has created an atmosphere directed toward slowly changing materials. Yet it should be noted that ``nothing in the nature of digital technology mandates that a digital library should include only rarely changing, long-lasting documents'' [33]. In general, document management systems do not deal well with versions or custom documents. Yet annotations, as a communication device, are expected to be somewhat transient. Versioning of both the reference and the annotation is critical in fluid environments to avoid skew, the condition resulting when the annotation refers to a different version of the reference document than currently available.
The desire for annotation of webpage content has resurfaced recently. Two products have emerged for accomplishing this task. Third Voice [25], is a Javascript plug-in for Internet Explorer which allows users to annotate webpages by depositing their public and group comments in a centralized annotation database. Alternatively, private annotations can be kept on their local machine. A second group is working on an open source method of annotation called Crit [60]. This method uses a mediation server to retrieve and combine a webpage with its available annotations. This server then presents to a regular web browser a ``value-added'' version of the original hypertext documents. As with the Third Voice model, annotations are kept in a centralized data repository.
In collaboration with Lincoln Stein, I propose to design and develop a distributed annotation system (DAS) which will improve the community's ability to annotate genomic data. With the recent completion of the C. elegans genome, annotation efforts in this community are accelerating rapidly. This makes the C. elegans data a good model for the new system.
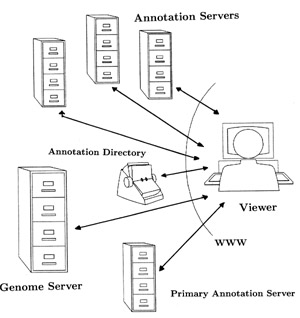
The DAS design is modeled after the current WWW. Web browsers are lightweight and are available on all platforms. Web pages are written in a simple standardized language, the hypertext markup language (HTML). This language can be produced in a text editor or by programs. A simple addressing scheme, uniform resource locators (URLs), identifies useful servers. Search engines and hot lists support the rapid location of relevant information. In the DAS design, a user would select from an annotation directory which annotation sources to view. Then the genome information and annotations would be combined in a ``layered'' fashion within the annotation viewer, much like layering transparencies. The principle change to the WWW design is the concept of multiple layers (URLs) integrated into a single view.
This system is best described as a multi-database system. Unlike federated and warehouse systems, this system requires a sematic-free schema rather than the stricter controlled vocabulary. Instead integration is achieved through a common display language. The system emphasizes the visualization of annotations rather than complex queries. The language will emphasize ``typesetting'' rather than the content of the document. In this way the system should be easily extensible to new annotation types.
The genome server is responsible for serving genome maps, sequences, and information related to the sequencing process. Initially there will be a single server, but mirror sites will be added as necessary.
Annotation servers are responsible for responding to requests on a region of a clone. A primary annotation server will be maintained by the sequencing center. Third party annotation servers can be built and maintained by any laboratory. Data sources need not have the same schema, but only communicate through a common language. This language will be XML (Extensible Markup Language) based. Versioning will be an integral part of the database reference, allowing for automatic handling of skew in most instances.
The directory access server is a small server maintained at the sequencing center that will provide clients with a list of current annotation servers. From this list, a client can select those annotation sources of personal interest.
The annotation viewer will be available in two separate versions, a lightweight and multi-platform stand alone application and a web based version. Both versions will allow users to browse the sequence and annotations and to pose simple queries. An important aspect of the viewer will be that there is no hard coded representation of the features. Instead, annotation types will be dynamically associated with graphical representations using a cascading style sheet [43] approach.
Subsequent developments will focus on moving from this ACeDB based initial prototype to a full application system. This will depend upon a XML based annotation language. The specification of this language is provided at http://biodas.org/documents/spec.html. It is still under development and revision.
| Scalability | Data sources will be distributed across the internet rather than residing in one centralized monolithic system. |
| Maintainability | Individual annotators will own and maintain their own layers, instead of having them curated by a central team. A pride of ownership will become the driving force for remaining current. |
| Improvement by Competition | ``Market forces'' will ensure that successful layers survive while unsuccessful and outdated layers disappear from the client's hotlist. |
| Specialization | The divide-and-conquer approach to annotation will permit experts to provide and maintain their own layers without the need to become computer experts. Specialized layers will develop to address individual communities. |
| Ease of Distribution | Because the client needs only a lightweight browser, local software installation and maintenance overhead will be minimized. |
| Quality Control | The end user's selection of layers provides the ultimate in selection and control over the annotations presented. Word of mouth and publication will become critical factors in determining who to trust for annotation. |
| Portability | The separation of sequence and map information from annotation allows them to be stored and represented in a variety of databases and schemas. |