Distributed Annotation System (DAS)
Version 1.6
October 19, 2010
Andrew M. Jenkinson, Jonathan Warren, Rafael C. Jimenez, Leyla J. Garcia, and the rest of the DAS community.
Edited by Andrew M. Jenkinson.
Based on
version 1.53 by Lincoln D. Stein, Sean Eddy and Robin Dowell.
This is a working document describing the protocol for a distributed sequence
annotation system. The original rationale is described in a
separate document.
Modifications introduced in version 1.6 are indicated in brown.
Deprecated content, indicated in pink, describes
behaviour that is no longer expected but must be tolerated by clients and
servers. It does not need to be acted on however. Deleted content, indicated
in blue, describes behaviour that is no longer
permitted. See the Changes
section for a full list of changes to the specification.
-
Description of the System
- Reference Objects
- Annotations
- Coordinate Systems
- Reference and Annotation Servers
- DAS Registry
- Clients
-
Client/Server Interactions
- The Request
- The Response
- Reference object IDs
-
Commands and Capabilities
- sources
- entry_points
- sequence
- types
- features
- stylesheet
- structure
- dsn [deprecated]
- dna [deprecated]
- link [deprecated]
- Exception Handling
- Fetching Sequence Assemblies
-
Feature Types and Categories
- Ontological Terms
- Categories
- Stylesheet Glyph Types
- Changes
This section provides a high-level view of the system architecture.
The Distributed Annotation System is a network of server and client software
installations distributed across the web. The DAS protocol is a standard
mechanism through which clients can communicate with servers in order to
obtain various types of biological data. The protocol defines:
- the communication method
- the query model
- the data format
By enforcing these constraints, DAS allows a client to integrate data from
many diverse sources implementing the protocol at a scaleable development
cost.
The DAS network of servers comprises a registry, several
reference servers and several annotation servers. Tying
these together are the concepts of reference objects and
coordinate systems.
Reference objects are items of data with stable identifiers that are targets
for annotation. At the most abstract level a reference object might be any
annotatable concept or idea, but usually describes a biological unit within
which annotations can be positioned. For example, "P15056" refers to a
protein sequence upon which annotations can be based. Similarly,
"chromosome 21" refers to a DNA sequence.
Individual reference objects can in fact have several versions, and it is
important to recognise that annotations based upon different versions of the
same reference entity are not necessarily equivalent.
Annotations are pieces of information that are attributed to a reference
object. Annotations are usually positional, that is they refer to
specific location within a reference object. An exon within a genomic
sequence is such an annotation. Annotations can also be
non-positional, in which case they can be considered as information
attributed to the whole of the reference object. For example, a textual
description of a protein or gene.
A coordinate system is a stable, logical set of reference objects. It
provides a mechanism to uniquely identify reference objects that share
identifiers, such as chromosomes. For example, chromosome 21 might identify
several reference objects from different species', but only one within the
NCBI 36 human assembly. Thus, "human NCBI 36 chromosomes" is a coordinate
system containing 25 reference objects (22 autosomes, X, Y and MT).
Coordinate systems are formally described using four properties:
- The category or type of object. For example a chromosome, contig or protein sequence.
- The authority responsible for defining the coordinate system. For example NCBI or UniProt.
- The version, for coordinate systems containing entities that are not versioned (e.g. genomic assemblies).
- The species, for coordinate systems containing only entities from a single organism.
Of these, category and authority are required properties. Version and species
are optional.
A full list of coordinate systems is available from the
DAS Registry.
Some examples are given below:
| Category | Authority | Version | Species |
|---|
| Chromosome | NCBI | 36 | Homo sapiens |
| Scaffold | ZFISH | 7 | Danio rerio |
| Protein sequence | UniProt | - | - |
Note: all properties of coordinate systems are case
sensitive.
A reference server is a DAS server that provides core data for the reference
objects in a particular coordinate system. For example, the reference server
for "UniProt Protein sequence" provides the actual sequence for each UniProt
entry. It does this by implementing the DAS sequence command. So
that clients can discover the available reference objects in a coordinate
system, a reference server must also list them via the entry_points
command. See the Queries section for further
details of these.
As it is responsible for providing "core data" for a coordinate system, a
reference server also defines the identifiers and versions that are used
across all servers using that coordinate system. For example, the reference
server for UniProt protein sequences is authoritative for the accessions that
may be used, and dictates that versions should be represented as MD5 checksums.
Annotation servers are specialised for returning lists of annotations for the
reference objects within a coordinate system. This is done by implementing
the DAS features command.
Note: The distinction between reference and annotation
servers is conceptual rather than physical. That is, a single server instance
can in fact play both roles by offering both sequences and annotations of
those sequences.
Note: A server may support multiple coordinate systems,
provided they do not contain reference objects with the same identifier.
The DAS Registry is a
special component of DAS, fulfilling the following roles:
- Catalogues and describes the capabilities and coordinate systems of DAS servers
- Allows discovery of available DAS services via both human and programmatic interfaces
- Automatically validates registered DAS sources to ensure that they correctly implement the protocol
- Periodically tests DAS sources and notifies their administrators if they are unavailable
- Provides a mechanism for activating or highlighting individual DAS services in clients
A DAS client typically integrates data from a number of DAS servers, making
use of the different data types. For example, a client might implement the
following procedure for a particular sequence location:
- Contact DAS registry to find reference and annotation servers for a particular genomic assembly
- Obtain sequence from the reference server
- Obtain sequence features from each of the annotation servers
- Display the annotations in the context of the sequence
This example is also available in
diagrammatic form.
The DAS is web-based. Clients query the reference and annotation servers
using the HTTP protocol (see
RFC2616) by sending a formatted URL request to the server. Servers
process the request and return a response in the form of a formatted XML
document (see W3C Extensible
Markup Language) according to a predefined schema.
All DAS requests take the form of a standard HTTP request with some DAS-specific
headers, submitted to a server using a hierarchical URL. Each URL has a
site-specific prefix, followed by a standardised path and query string. The
standardised path begins with the string /das. This is
followed by URL components containing the data source name
and/or a command. The following are examples of valid
DAS requests:
http://www.ebi.ac.uk/das-srv/genomicdas/das/sources
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ ^^^ ^^^^^^^
site-specific prefix das command
In this case, the site-specific prefix is http://www.ebi.ac.uk/das-srv/genomicdas. The
request begins with the standardised path /das. This is followed by
the command /sources.
http://das.sanger.ac.uk/das/ccds_mouse/features?segment=1:174405453,174408689
^^^^^^^^^^^^^^^^^^^^^^^ ^^^ ^^^^^^^^^^ ^^^^^^^^ ^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
site-specific prefix das data src command arguments
In this case, the site-specific prefix is http://das.sanger.ac.uk. The
request begins with the standardised path /das and the data source, in
this case /ccds_mouse. This is followed by the command /features,
and a query string providing named arguments to the features command.
Thus, a single DAS server hosts one or more DAS data sources,
allowing it to provide information from different projects or for several
coordinate systems. Each server as a whole, and each data source individually,
supports one or more DAS commands (or requests),
allowing it to respond to different types of query. In the first example,
the EBI server will provide a list of data sources via the sources command.
In the second example, the ccds_mouse data source on the Sanger server
will provide a list of annotations via the features command. The same
server in fact provides a number of other data sources, and each responds
to its own set of commands. Whether the request is for a server command or a
source command is known as its scope.
More information on the format of the request and the various available
commands is given in the Queries section.
Data Source Names
Previous versions of the DAS specification required that the data source
name be a standard identifier for a genome assembly and thus was
identical across servers annotating the same reference genome. This
restriction no longer applies. Instead, it is recommended for a DSN to
identify the reference coordinates and origin/type of the data. For genomic
sources, the recommended format is genome|track
(e.g. hg18|refseq).
Command Arguments
The query string portion of the request (the "?" symbol rightward) can be
submitted to the URL following conventional HTTP standards, using either the
GET or POST methods. For large queries, POST is recommended.
Request Headers
In each request, a DAS client should supply a standard HTTP header, containing
within it information about the DAS client:
- X-DAS-Version - the protocol version (currently "DAS/1.6")
- X-DAS-Client - the client name and version
Clients may also implement the Cross-Origin
Resource Sharing extension to the HTTP specification. This provides a robust
mechanism for javascript DAS clients implemented in web browsers to perform
cross-site requests in order to access remote DAS servers.
Example
This example is a Cross-Origin request from an client at ebi.ac.uk to
a server at sanger.ac.uk. The client is requesting annotations for a
segment of chromosome 1 from the ccds_mouse data source using the
features command.
GET /das/ccds_mouse/features?segment=1:174405453,174408689 HTTP/1.1
Host: das.sanger.ac.uk
User-Agent: Bio::Das::Lite/1.2 libwww-perl/5.814
Origin: http://www.ebi.ac.uk
X-DAS-Client: Dasty/3.0
X-DAS-Version: DAS/1.6
The response from the server to the client consists of a HTTP header
with DAS-specific information within that header, followed optionally by XML
content that contains the answer to the query. The DAS-specific portion of the
header consists of four lines:
- X-DAS-Version - the protocol version (currently "DAS/1.6")
- X-DAS-Status - a three digit status code which indicates the outcome of the request
- X-DAS-Capabilities - describes the parts of of the specification the server implements
- X-DAS-Server - the server name and version
Servers must also respond appropriately to a Cross-Origin
Resource Sharing request if a client makes one. Although server implementors
may choose not to, it is recommended that servers adopt an all-origins policy.
This allows browser-implemented DAS clients to function on a par with those
without such cross-site restrictions.
Here is an example HTTP header (provided by DAS server):
HTTP/1.1 200 OK
Date: Sun, 12 Mar 2000 16:13:51 GMT
Last-Modified: Fri, 16 Feb 2009 11:17:59 GMT
Content-Type: text/xml
Access-Control-Allow-Origin: *
Access-Control-Expose-Headers: X-DAS-Version, X-DAS-Status, X-DAS-Capabilities, X-DAS-Server
X-DAS-Version: DAS/1.6
X-DAS-Status: 200
X-DAS-Capabilities: error-segment/1.0; unknown-segment/1.0; unknown-feature/1.0; ...
X-DAS-Server: ProServer/553
data follows...
Status Codes
This aspect of the protocol has been clarified in this version of the
specification.
A DAS server must provide an X-DAS-Status code in addition to a HTTP
status code. HTTP status codes indicate the status of the request as defined
by the HTTP/1.1 specification, whereas X-DAS-Status indicates the status of
the request as it pertains to DAS. In this manner, the HTTP status reporting
mechanism is not compromised through use of DAS - it is instead enhanced by
more specific reporting of why a request has failed. This allows other HTTP
features such as redirection and authentication to be included within DAS, and
it is therefore important for implementations to interpret the errors
correctly. DAS clients should inspect both codes to be sure to catch all
possible errors.
The defined X-DAS-Status codes are listed in Table 1. For example, when a DAS
client makes a request for a data source that does not exist on the server,
the appropriate DAS error code is "401 Bad data source". This is a more
specific error condition than the generic HTTP "400 Bad request" status. Note
that the HTTP status "401 Unauthorized" is not to be used
to indicate this error condition. Note also the difference between DAS errors
400 and 501 - the former indicates that the client is requesting an invalid
command (a client error); the latter indicates the command is valid but the
data source does not implement it (a server error).
Table 1: DAS response codes
| HTTP Status | HTTP Description | X-DAS-Status | Description |
|---|
| 200 | OK, data follows | 200 | OK, data follows |
|---|
| 400 | Bad Request | 400 | Bad command (command not recognized) |
|---|
| 400 | Bad Request | 401 | Bad data source (data source unknown) |
|---|
| 400 | Bad Request | 402 | Bad command arguments (arguments invalid) |
|---|
| 404 | Not found | 404 | Bad stylesheet (requested stylesheet unknown) |
|---|
| 500 | Server error | 500 | Server error, not otherwise specified |
|---|
| 500 | Server error | 501 | Data source does not implement feature/command |
|---|
| 400 | Bad Request | 403 | Bad reference object (reference sequence
unknown) [deprecated in favour of Exception Handling] |
|---|
| 400 | Bad Request | 405 | Coordinate error (sequence coordinate is out
of bounds/invalid) [deprecated in favour of Exception Handling] |
|---|
This aspect of the protocol has been clarified in this version of the
specification.
All DAS responses must include a list of
the capabilities that the server or data source provides. A capability is either a
DAS command or an optional component of a
command. Note therefore that a command is not the same as a capability (all
commands are capabilities, but not all capabilities are commands).
This system allows clients to determine which functionality to expect from a
server. This includes the commands the server or data source will respond to, and whether
it conforms to any optional special behaviours for that command. It also allows for
future extensions to the DAS protocol to be reported by servers that support
them.
Capabilities are primarily reported by the X-DAS-Capabilities
HTTP header, which takes the form:
CapabilityName/Version; CapabilityName/Version
That is:
capabilityA/1.0; capabilityB/1.4; capabilityC/1.0
The version of each capability is an alternative method for clients
to determine compatibility between specification versions. It tracks which
capabilities have changed from one DAS specification version to the next.
All servers must report the correct capability versions for the version of the
specification they support. For example, the features command is version 1.1
in DAS version 1.6. Servers may NOT mix capability versions from different
specifications, or otherwise vary the capability version without varying the
DAS version.
The capabilities a server reports will depend on
the scope of the request. That is, if a client issues a server-level
command such as sources, the list of capabilities will be limited to
the supported server-level commands. If the scope of the request is for a
specific data source (such as types), the list of capabilities
will be limited to its supported source-level commands. A source should not
report the capabilities of its server.
For example:
<SERVER>/das/sources
X-DAS-Capabilities: sources/1.0; dsn/1.0
<SERVER>/das/<DSN>/types
X-DAS-Capabilities: features/1.1; types/1.1; stylesheet/1.1; unknown-segment/1.0; maxbins/1.0
Capabilities are also reported in the body of the response to the sources
command, described later.
The following standard capabilities are present in the DAS/1.6 protocol:
| Capability Name | Description |
| dsn/1.0 |
The server supports the deprecated dsn request.
|
| dna/1.0 |
The dna request is no longer valid.
|
| sequence/1.1 |
The server supports the basic sequence request.
|
| types/1.1 |
The server supports the basic types request.
|
| stylesheet/1.1 |
The server supports the basic stylesheet request.
|
| features/1.1 |
The server supports the basic features request.
|
| entry_points/1.1 |
The server supports the basic entry_points request.
|
| sources/1.0 |
The server supports the basic sources request.
|
| structure/1.0 |
The server supports the basic structure request.
|
| error-segment/1.0 |
Server will report requests for invalid segments with an
<ErrorSegment> response.
|
| unknown-segment/1.0 |
Server will report requests for unknown or unannotated segments with an
<UnknownSegment> response.
|
| unknown-feature/1.0 |
Server will report requests for unknown features with an
<UnknownFeature> response.
|
| feature-by-id/1.0 |
The features request will accept the CGI parameter "feature_id", enabling
the server to look up segment(s) based on the ID of a feature.
|
| group-by-id/1.0 |
The features request will accept the CGI parameter "group_id", enabling
the server to look up segment(s) based on the ID of a group of features.
|
| component/1.0 |
The features request will return components of the indicated segment when
a category type of "component" is requested.
|
| supercomponent/1.0 |
The features request will return supercomponents of the indicated segment when
a category type of "supercomponent" is requested.
|
| maxbins/1.0 |
The features request will result in different sets of features,
depending on the client's available rendering space.
|
Content Type
The response to all successful DAS commands takes the form of an XML document
that is not intended for human consumption. As such, the appropriate content
type (set via the Content-Type HTTP header) is application/xml.
Servers should use this content type in all cases where it is supported by the
client (supported content types are reported by the client in the
Accept request header, as defined in the
HTTP specification).
If the client only supports text/xml, the server may use this
instead.
Compression
The HTTP/1.1 protocol allows web clients to request byte-level compression of
the response by sending the Accept-Encoding HTTP header.
Web servers that are capable of it can reply with a Content-Encoding
header and a compressed body. Implementors of DAS clients and servers may
wish to implement this HTTP feature.
The ID used by a client or server to refer to a reference object can contain
any set of printable characters (including the space character),
except for the following characters:
- the colon character (":"), which is reserved for separating reference IDs from sequence ranges (see below).
- the semicolon (";") and ampersand ("&") characters, which are reserved for separating request parameters.
A data source that uses a reserved character in its internal IDs must apply a
mapping on the way in and on the way out. For example:
Client request Server's internal id Response to client
gi-123456 --> gi:123456 ---> gi-123456
gi-123456:1,1000 --> gi:123456 start=1 stop=1000 ---> gi-123456:1,1000
MGI12345 --> MGI:12345 ---> MGI12345
Several parts of this document refer to segments. These are
regions of reference objects. Depending on the context, a segment
may be an individual reference object (so called because genome assemblies
are usually divided into distinct parts), or a region within a single
reference object. The difference has negligible effect on the interpretation
of the documentation.
Where used as query parameters, a segment is always formatted as follows:
id:start,end
For example, a 100 kb region of chromosome X:
X:200001,300000
This section lists the queries recognised by reference and annotation servers.
As described in the Request section, each of these
contains a site-specific prefix, denoted here as SERVER, and usually
a data source name, denoted here as DSN. Where a server supports a
command, it must be reported as a capability.
Description:
This query returns the list of data sources that are available from this server,
along with additional metadata to describe each source's capabilities.
Scope:
DAS Registry, reference servers and annotation servers. It is required
for all.
Request:
This command is executed relative to the server, in one of two ways.
1. To list all sources:
SERVER/das/sources
2. To limit the response to a single data source:
SERVER/das/DSN
Arguments:
The DAS Registry implements a more advanced form of the sources
command allowing the list of sources to be filtered. It supports the following
named arguments (applicable to the first URL format only):
- capability (optional; zero or one)
- Limits the list of sources to those that support the given capability
(e.g. "features").
- type (optional; zero or one)
- Limits the list of sources to those that support a
coordinate system of the given reference object type. For
example, "Chromosome" or "Protein Sequence"
- authority (optional; zero or one)
- Limits the list of sources to those that support a
coordinate system of the given authority (e.g. "NCBI").
- version (optional; zero or one)
- Limits the list of sources to those that support a
coordinate system of the given version (e.g. "36").
- organism (optional; zero or one)
- Limits the list of sources to those that support a
coordinate system of the given species. The species may be a taxonomy
code or full name.
- Note that sources with coordinate systems that are not species-specific
(i.e. support all species') will not be returned if this filter is specified.
- label (optional; zero or one)
- Limits the list of sources to those that are labelled with the given string.
Note: Combining parameters of different types is treated as a
logical AND (intersection) operation. It is not possible to combine parameters
of the same type.
Arguments Example:
This URL requests a list of data sources which offer the capability sequence,
for Chromosome reference objects in the human species.
http://www.dasregistry.org/das/sources?capability=sequence&organism=9606&type=Chromosome
Response:
The response to the sources command is the "SOURCES" XML-formatted document:
<?xml version='1.0' standalone="no" ?>
<?xml-stylesheet type="text/xsl" href="das.xsl"?>
<SOURCES>
<SOURCE uri="URI" title="title" doc_href="helpURL" description="description">
<MAINTAINER email="email address" />
<VERSION uri="URI" created="date">
<COORDINATES uri="URI"
source="data type"
authority="authority"
taxid="taxonomy"
version="version"
test_range="id:start,stop" >coordinate string</COORDINATES>
<CAPABILITY type="das1:command" query_uri="URL" />
<PROP name="key" value="value" />
</VERSION>
<VERSION ...>
...
</VERSION>
</SOURCE>
<SOURCE ...>
...
</SOURCE>
</SOURCES>
The response XML is formally described by a RELAX NG schema definition,
and is explained below:
- <SOURCES> (required; one only)
- The appropriate root tag is SOURCES.
- <SOURCE> (optional; zero or more)
- There are zero or more <SOURCE> tags, each of which represents one
dataset.
- The uri (required) attribute uniquely identifies the source, and
must be globally unique amongst all DAS sources. See the URIs section below
for more details.
- The title (required) attribute is a short text label suitable for
display, and the description (required) attribute is a longer text
description. Neither may contain markup.
- The doc_href (optional) attribute is a URL location where more
information about the data is available. The target may be any
browser-readable MIME-type.
- <MAINTAINER> (required; one per SOURCE tag)
- The maintainer tag identifies the contact person for the source.
- The email (required) attribute is a properly formatted email address.
- <VERSION> (required; one or more per SOURCE tag)
- This tag represents one independent version of a data source.
- The uri (required) attribute is a global unique identifier for this
version of the data source. See the URIs section below
for more details.
- In this version of DAS, there is only one VERSION per SOURCE and the version
URI is the same as the source URI. In future,
multiple versions may be permitted to allow servers to signal to clients that
there is a later version of a set of data without affecting those reliant
on the old set. The version URI should therefore be considered the
unique identifier for a queryable DAS source.
- The created (required) attribute is the
publish date in ISO 8601 format as adopted by the W3C.
- <COORDINATES> (required; one or more per VERSION tag)
- This tag identifies a reference coordinate system
supported by the source.
- The uri (required) attribute is a globally unique identifier for
the coordinate system. It should be a fully resolvable URL providing more
information about the coordinate system.
- The URI of a coordinate system is defined by the DAS Registry.
- The authority (required) attribute is the
project or organisation responsible for defining the coordinate system
(e.g. NCBI or UniProt).
- The source (required) attribute is the
type of reference object (e.g. "Chromosome" or "Protein Sequence").
- The test_range (required) attribute is an example
segment that may be used to test the source's
capabilities. It should be a segment for which the source has data.
- The taxid (optional) attribute is the
NCBI taxonomy ID for
the species, where a coordinate system is restricted to only one species.
- The version (optional) attribute is used to indicate the
version of a coordinate system as a whole. It is used to differentiate
between coordinate systems that change infrequently but are fundamentally
incompatible between releases. Typically these are genomic.
- The content of the COORDINATES tag is a string representation of the
coordinate system: authority[_version],source[,species full name]
- <CAPABILITY> (required; one or more per VERSION tag)
- This tag describes the commands and other capabilities supported by the
data source.
- The type (required) attribute is a formatted string of the format:
das1:capability_name
For example: "das1:entry_points" or
"das1:feature-by-id". Capability names must match those described in the
capabilities section.
- The "das1:" prefix allows servers and clients to distinguish DAS
capabilities from any that are not related to DAS. It is thus for future use.
- The query_uri attribute is the actual DAS URL used to execute a
capability. It is required for capabilties that are commands
(e.g. "features") and does not apply to other capabilities (e.g.
"feature-by-id").
- The sources command is mandatory for all sources, and the URI must
point to the URL of the source on its server. See the examples below for details.
- Whilst in future it may be possible to specify query_uris for
different capabilities independently, currently they must all follow the
DAS standard URL construction rules and refer to the same data source.
See the examples below for more details.
- <PROP> (optional; zero or more per VERSION tag)
- This tag allows a source to be annotated with custom properties. It has
name (required) and value (required) attributes. Some common
properties are set by the DAS Registry.
The URI (Uniform Resource Identifier) of a DAS data source is a
unique identifier on the web. It is also a URL (Uniform Resource Locator),
which means it must also be a fully resolvable web document. A DAS source's
URI/URL is the location of a sources document
describing the data source. At the most basic level, this URL would be the full
DAS URL for the source on its server, i.e.:
SERVER/das/DSN (e.g. http://www.example.com/das/mysource)
When it is included in a sources document, a URL can be either
relative or absolute in the same way as an HTML document.
In DAS, a relative URL is used when referring to a source on the same
server as the HTTP request, whereas an absolute URL is used when referring to
a location on a different server.
This is important as the same source may be listed in sources
responses from different servers - specifically, the server hosting the source
and the DAS Registry.
When a source is listed in the DAS Registry, the Registry assigns a URI in its
own namespace (e.g. http://www.dasregistry.org/das/DS_1234). This URI is
independent of the location of the server hosting the source, allowing the
source to move if necessary. The server hosting the source should use the
Registry URL to refer to the source in its own sources response,
allowing clients to determine that the two entries are the same.
For example, responses to both of these queries might refer to the
same DAS source:
http://www.example.com/das/mysource
http://www.dasregistry.org/das/DS_1234
It is expected that these documents remain in sync with each other, and refer
to the source using the same version URI. Thus the server maintainer
should endeavour to ensure that DAS Registry absolute URLs are used in the
server's sources document.
In addition, the current "home server location" of the data source is always
provided via the query URI for the sources capability.
Sources Command Examples
The following examples illustrate various potential
combinations for a response to the sources command.
Example 1
This is a request direct to a server hosting a source that is not registered
in the DAS Registry. Note that because the sources command is
mandatory, the sources response can always be retrieved via a GET
request using the data source's version URL:
GET http://www.example.com/das/transcripts
<?xml version="1.0"?>
<SOURCES>
<SOURCE uri="transcripts"
title="Example Transcripts"
description="Examples of transcripts in the human genome">
<MAINTAINER email="person@example.com" />
<VERSION uri="transcripts" created="2010-06-16T11:53:29+0000">
<COORDINATES uri="http://www.dasregistry.org/dasregistry/coordsys/CS_DS311"
taxid="9606"
source="Chromosome"
authority="GRCh" version="37"
test_range="4:32211548,32711547">GRCh_37,Chromosome,Homo sapiens</COORDINATES>
<CAPABILITY type="das1:sources" query_uri="http://www.example.com/das/transcripts" />
<CAPABILITY type="das1:features" query_uri="http://www.example.com/das/transcripts/features" />
</VERSION>
</SOURCE>
</SOURCES>
Note the use of relative and absolute URLs (the highlighted components), and that the source and
version URIs are both the same.
Example 2
The next example illustrates a sources response from a request for a specific
source in the DAS Registry. Here, the version URI
is a globally unique identifier for the source within the Registry's namespace.
GET http://www.dasregistry.org/das/DS_566
<?xml version="1.0"?>
<SOURCES>
<SOURCE uri="DS_566"
title="GRC regions"
doc_href="http://www.sanger.ac.uk/sequencing/grc/"
description="Regions reported to the Genome Reference Consortium">
<MAINTAINER email="person@institute.ac.uk" />
<VERSION uri="DS_566" created="2008-06-10T20:37:09+0000">
<COORDINATES uri="http://www.dasregistry.org/dasregistry/coordsys/CS_DS40"
taxid="9606"
source="Chromosome"
authority="NCBI" version="36"
test_range="4:32211548,32711547">NCBI_36,Chromosome,Homo sapiens</COORDINATES>
<CAPABILITY type="das1:sources" query_uri="http://das.sanger.ac.uk/das/grc_region" />
<CAPABILITY type="das1:features" query_uri="http://das.sanger.ac.uk/das/grc_region/features" />
<CAPABILITY type="das1:stylesheet" query_uri="http://das.sanger.ac.uk/das/grc_region/stylesheet" />
<PROP name="leaseTime" value="2009-02-25T07:47:47+0000" />
</VERSION>
</SOURCE>
</SOURCES>
Note that the URI is independent of the location of the DAS server itself,
allowing clients to adapt to server moves. The location of the source on the
DAS server is however provided in the query URI for the das1:sources
capability.
Example 3
The same information as above can also be retrieved direct from the DAS server
itself:
GET http://das.sanger.ac.uk/das/grc_region
<?xml version="1.0"?>
<SOURCES>
<SOURCE uri="http://www.dasregistry.org/das/DS_566"
title="GRC regions"
doc_href="http://www.sanger.ac.uk/sequencing/grc/"
description="Regions reported to the Genome Reference Consortium">
<MAINTAINER email="person@institute.ac.uk" />
<VERSION uri="http://www.dasregistry.org/das/DS_566" created="2008-06-10T20:37:09+0000">
<COORDINATES uri="http://www.dasregistry.org/dasregistry/coordsys/CS_DS40"
taxid="9606"
source="Chromosome"
authority="NCBI" version="36"
test_range="4:32211548,32711547">NCBI_36,Chromosome,Homo sapiens</COORDINATES>
<CAPABILITY type="das1:sources" query_uri="http://das.sanger.ac.uk/das/grc_region" />
<CAPABILITY type="das1:features" query_uri="http://das.sanger.ac.uk/das/grc_region/features" />
<CAPABILITY type="das1:stylesheet" query_uri="http://das.sanger.ac.uk/das/grc_region/stylesheet" />
<PROP name="leaseTime" value="2009-02-25T07:47:47+0000" />
</VERSION>
</SOURCE>
</SOURCES>
Note that the version URI for this source is not relative to the server, but
because the source is registered uses its Registry URL instead.
Description:
This query returns the list of reference objects known by a data source. These
serve as entry points for other commands. In the case of genomic assemblies,
the "top level" reference objects are listed.
Scope:
Reference and annotation servers. This command is
required for reference servers.
Request:
This command is executed relative to a data source:
SERVER/das/DSN/entry_points[?rows=start-end]
Arguments:
- rows (optional)
- Limits the entry points returned in the response to those in the given
range, allowing the client to retrieve a smaller cross-section of the
results at any one time. This is particularly important for coordinate
systems with large numbers of entry points (such as UniProt).
The parameter takes the form start-end.
- If omitted, it is up to the server to select a suitable default. Omitting
the parameter is therefore not guaranteed to return all entry points - a
server may choose to only return the first few, for example.
- In addition, the server is free to return only a subset of the requested
rows if it deems the requested range to be too large.
Response:
The response to the entry_points command is the "DASEP" XML-formatted document:
<?xml version="1.0" standalone="no"?>
<DASEP>
<ENTRY_POINTS href="url" total="total entry points" start="first in list" end="last in list">
<SEGMENT id="id1" start="start1" stop="stop1" version="X.XX" orientation="+">label1</SEGMENT>
<SEGMENT id="id2" start="start2" stop="stop2" version="X.XX" orientation="+">label2</SEGMENT>
<SEGMENT id="id3" start="start3" stop="stop3" version="X.XX" orientation="+">label3</SEGMENT>
...
</ENTRY_POINTS>
</DASEP>
The response XML is formally described by a RELAX NG schema definition, and is
explained below:
- <!DOCTYPE> (required; one only)
- The doctype indicates which formal DTD specification to use.
For the entry_points query, the doctype DTD is "http://www.biodas.org/dtd/dasep.dtd".
- <DASEP> (required, one only)
- The appropriate root tag is DASEP.
- <ENTRY_POINTS> (required, only one)
- There is a single <ENTRY_POINTS> tag.
- The version
attribute is in the form "N.NN". Whenever the
sequence of an entry point changes, the version number should change as well.
- The href (required) attribute echoes the URL query that
was used to fetch the current document.
- The total (required) attribute is the total number of entry points
known by the source, irrespective of how many are returned in the current
response.
- The start and end (optional) attributes indicate
where the returned entry points lie in the total list available from the
server. Typically, these will echo the rows parameter, but this is
not mandatory: the server can elect to return a subset of the requested
rows if it wishes and, when the rows parameter is omitted, the server
chooses its own defaults.
- <SEGMENT> (optional; zero or more)
- Each entry point is represented by a segment element.
- The id is a unique identifier for a reference object, and can be used as
the reference ID in further requests to DAS.
- The start and stop attributes indicate the size of the segment.
The server must include these if it knows them, but they are otherwise optional.
- The version attribute (optional) indicates the version
of the reference object, used for coordinate systems which are not
themselves versioned.
- The optional orientation attribute indicates the strandedness of
the segment. Its value is one of "+" or "-" or "0". If omitted, a value of
"0" (no intrinsic strand) is assumed.
- If the optional subparts attribute is present and has the value "yes",
it indicates that the segment has subparts.
- If the optional type attribute is present, it can be used to describe the
type of the reference object. It should match the type component
of the coordinate system.
- The tag content (optional) provides a human readable for display purposes.
If omitted, it is assumed the ID is appropriate for display.
- For compatibility with older versions of the specification, the <SEGMENT>
tag can use a size attribute rather than start and stop:
<SEGMENT id="id" size="123456">
In this case, the start is assumed to be "1" and the stop is assumed to be the same
as the length.
Note: The result from the entry points requests only lists top level
segments, and thus does not carry sufficient information to reconstruct a
complex sequence assembly. Instead, use the features request with a category
of "component". See Fetching Sequence Assemblies.
Note: The ability to limit results to a cross section of the total list
of entry points requires that entry points always be returned in the same
order. Though the exact ordering strategy is left to the server, it is
recommended to sort by object type and lexographically.
Note: If a client requests an invalid range of rows (e.g. completely
beyond the range offered by the server, or negative values) the server
responds with an X-DAS-Status of 402 (see the Request
section). However, if the server has entry points for at least one of the
requested range of rows, they are returned as normal (the actual rows returned
being indicated via the start and end attributes).
Examples
Here is an example of a valid request that uses the rows argument to
retrieve only 10 entry points, starting at the 11th:
http://www.ebi.ac.uk/das-srv/uniprot/das/uniprot/entry_points?rows=11-20
Here is an example of a request and its response:
http://www.ensembl.org/das/Homo_sapiens.GRCh37.reference/entry_points?rows=21-29
<DASEP>
<ENTRY_POINTS href="http://www.ensembl.org/das/Homo_sapiens.GRCh37.reference/entry_points" total="93" start="21" end="29">
<SEGMENT type="Chromosome" id="8" start="1" stop="146364022" orientation="+" subparts="yes">8</SEGMENT>
<SEGMENT type="Chromosome" id="9" start="1" stop="141213431" orientation="+" subparts="yes">9</SEGMENT>
<SEGMENT type="Chromosome" id="MT" start="1" stop="16569" orientation="+" subparts="yes">MT</SEGMENT>
<SEGMENT type="Chromosome" id="X" start="1" stop="155270560" orientation="+" subparts="yes">X</SEGMENT>
<SEGMENT type="Chromosome" id="Y" start="1" stop="59373566" orientation="+" subparts="yes">Y</SEGMENT>
<SEGMENT type="Supercontig" id="GL000191.1" start="1" stop="106433" orientation="+" subparts="yes">GL000191.1</SEGMENT>
<SEGMENT type="Supercontig" id="GL000192.1" start="1" stop="547496" orientation="+" subparts="yes">GL000192.1</SEGMENT>
<SEGMENT type="Supercontig" id="GL000193.1" start="1" stop="189789" orientation="+" subparts="yes">GL000193.1</SEGMENT>
<SEGMENT type="Supercontig" id="GL000194.1" start="1" stop="191469" orientation="+" subparts="yes">GL000194.1</SEGMENT>
</ENTRY_POINTS>
</DASEP>
Description:
This query returns the sequence (nucleotide or protein) corresponding to the
indicated segment.
Scope:
Reference servers.
Request:
This command is executed relative to a data source:
SERVER/das/DSN/sequence?segment=RANGE[;segment=RANGE...]
Arguments:
- segment (required; one or more)
- Each segment argument uses a format of either
reference:start,stop or reference, where
reference is the ID of the reference object and start and stop are the endpoints of
the region of sequence requested, inclusive.
- If the start and stop positions are not provided, they default to the endpoints
of the entire reference sequence. If start and stop are provided at least one
must be a valid position within the reference object, and start may not be
less than 1 or greater than end.
Response:
The response to a sequence request is the "DASSEQUENCE" XML-formatted document:
<?xml version="1.0" standalone="no"?>
<DASSEQUENCE>
<SEQUENCE id="id" start="start" stop="stop" version="X.XX" label="Label">
atttcttggcgtaaataagagtctcaatgagactctcagaagaaaattgataaatattat
taatgatataataataatcttgttgatccgttctatctccagacgattttcctagtctcc
agtcgattttgcgctgaaaatgggatatttaatggaattgtttttgtttttattaataaa
taggaataaatttacgaaaatcacaaaattttcaataaaaaacaccaaaaaaaagagaaa
aaatgagaaaaatcgacgaaaatcggtataaaatcaaataaaaatagaaggaaaatattc
agctcgtaaacccacacgtgcggcacggtttcgtgggcggggcgtctctgccgggaaaat
tttgcgtttaaaaactcacatataggcatccaatggattttcggattttaaaaattaata
taaaatcagggaaatttttttaaattttttcacatcgatattcggtatcaggggcaaaat
tagagtcagaaacatatatttccccacaaactctactccccctttaaacaaagcaaagag
cgatactcattgcctgtagcctctatattatgccttatgggaatgcatttgattgtttcc
gcatattgtttacaaccatttatacaacatgtgacgtagacgcactgggcggttgtaaaa
cctgacagaaagaattggtcccgtcatctactttctgattttttggaaaatatgtacaat
gtcgtccagtattctattccttctcggcgatttggccaagttattcaaacacgtataaat
aaaaatcaataaagctaggaaaatattttcagccatcacaaagtttcgtcagccttgtta
tgtcaaccactttttatacaaattatataaccagaaatactattaaataagtatttgtat
gaaacaatgaacactattataacattttcagaaaatgtagtatttaagcgaaggtagtgc
acatcaaggccgtcaaacggaaaaatttttgcaagaatca
</SEQUENCE>
</DASSEQUENCE>
The response XML is formally described by a RELAX NG schema definition, and is
explained below:
- <!DOCTYPE> (required; one only)
- The doctype indicates which formal DTD specification to use.
For the sequence query, the doctype DTD is "http://www.biodas.org/dtd/dassequence.dtd".
- <DASSEQUENCE> (required; one only)
- The appropriate doctype and root tag is DASSEQUENCE.
- <SEQUENCE> (required; one or more)
- There is a single <SEQUENCE> tag per requested segment. It has the
attributes id, which indicates the reference ID for this sequence,
start and stop, which indicate the position of
this segment within the reference sequence. All are required.
- The version attribute (optional) indicates
the version of the reference object, used for coordinate systems which are
not themselves versioned.
- The label attribute (optional) supplies a human readable label
for display purposes. If omitted, it is assumed the ID is suitable for
display.
- The moltype attribute indicates the molecular type of
the sequence, being one of DNA, ssRNA, dsRNA, or Protein. No
provision is made for circular molecules. This attribute is deprecated,
being implicit in the coordinate system.
- The content of this tag is the sequence itself, using standard
IUPAC codes for DNA, RNA and protein.
Note: Servers implementing the sequence command are
required to also implement the error-segment
capability. See the Exception Handling section for
more details.
Examples
Here is an example of a valid request that uses the segment argument to
fetch three independent segments. The last two segments are subsequences:
http://www.ensembl.org/das/Homo_sapiens.GRCh37.reference/sequence?segment=Y;segment=X:1,1000;segment=1:50,200
Here is an example of an request with three invalid segment
arguments. The first because either both or neither start/end must be provided,
the second because start is an invalid position, and the third because
neither start nor end is within the reference object:
http://www.ensembl.org/das/Homo_sapiens.GRCh37.reference/sequence?segment=X:200;segment=Y:0,1000;segment=21:100000000,100001000
Description:
This query returns the types of annotation available for a data source.
Scope:
Annotation and reference servers. It is required for sources
implementing the features command.
Request:
This command is executed relative to a data source:
SERVER/das/DSN/types[?segment=RANGE]
[;segment=RANGE...]
Arguments:
- segment (optional)
- If provided, each segment argument uses a format of either
reference:start,stop or reference, where
reference is the ID of the reference object and start and stop are the endpoints of
the region to query, inclusive.
- If the start and stop positions are not provided, they default to the endpoints
of the entire reference sequence. If start and stop are provided at least one
must be a valid position within the reference object, and start may not be
less than 1 or greater than end.
- type (optional)
- One or more type IDs to be used for filtering annotations on the type
field. If multiple type IDs are provided, the resulting list of features
will be the logical OR of the list.
If one or more segment arguments are provided, the server returns a distinct
list of types for the features overlapping those segments (i.e. the features
that would be provided for the equivalent features query). If no segment
argument is provided, then all feature types known to the source are
returned.
Response:
The document returned from the types request is an
XML-formatted "DASTYPES" document. This is a shortened form of the
full features format (see below) and is used to summarise the type and
number of each annotation. Annotation types can be grouped into
segments, or be totaled across the entire database.
<?xml version="1.0" standalone="no"?>
<DASTYPES>
<GFF href="url">
<SEGMENT id="id" start="start" stop="stop" version="X.XX" label="label">
<TYPE id="id1" cvId="term1" category="category">Type Count 1</TYPE>
<TYPE id="id2" cvId="term2" category="category">Type Count 2</TYPE>
...
</SEGMENT>
</GFF>
</DASTYPES>
The response XML is formally described by a RELAX NG schema definition, and is
explained below:
- <!DOCTYPE> (required; one only)
- The doctype indicates which formal DTD specification to use.
For the types query, the doctype DTD is "http://www.biodas.org/dtd/dastypes.dtd".
- <DASTYPES> (required; one only)
- The appropriate root tag is DASTYPES.
- <GFF> (required; one only)
- There is a single <GFF> tag. The href (required) attribute
echoes the URL query that was used to fetch the current document.
- The version attribute indicates the current version of the XML form
of the General Feature Format.
- <SEGMENT> (required; one or more)
- The <SEGMENT> tag identifies the reference segment to which the list
of types applies.
- If no segment parameter was used in the request, the list of types applies
to the whole data source and this element has no attributes.
- Otherwise, the list of types applies to the specific query segment identified
by the required id attribute. The start and stop attributes
must be included if the server knows them or they can be
deduced from the request, but they are otherwise optional.
- If the segment corresponds to a defined region of
the genome. If the list of types corresponds to the entire database, these are optional.
- The version attribute (optional) indicates
the version of the reference object being annotated, used for coordinate systems which are
not themselves versioned.
- The label attribute (optional) supplies a human readable label
for display purposes. If omitted, it is assumed the ID is suitable for
display.
- The type attribute (optional) describes the reference
object type.
- <TYPE> (optional; zero or more per SEGMENT)
- Each segment has zero or more <TYPE> tags, which summarise
the types of annotation available.
- The id attribute (required) is an identifier for the annotation
type that is unique within the data source.
- The method attribute (optional) indicates the method
used to generate this type of feature. Since this can be independent of
the type, this is now deprecated.
- The category attribute (optional) provides functional
grouping to related types.
- The cvId attribute (optional, recommended) is the ID
of a term from a relevant controlled vocabulary (SO, MOD, BS). See the
Ontology section for more details.
- The tag contents (optional) is
a count of the number of features of this type across the segment.
Note: The contents of the <TYPE> elements must match
those provided in the response to the Features command.
Description:
This query returns the annotations available for a reference segment.
Scope:
Reference and annotation servers. It is required for sources
implementing the types command.
Request:
This command is executed relative to a data source:
SERVER/das/DSN/features?segment=RANGE
[;segment=RANGE]
[;type=TYPE]
[;type=TYPE]
[;category=CATEGORY]
[;category=CATEGORY]
[;feature_id=ID]
[;maxbins=BINS]
Arguments:
The features command accepts several argument types. Although all are
optional, at least one of either segment or feature_id must
be provided.
- segment (zero or more)
- If specified, the segment argument restricts the list of annotations to those that
overlap the indicated range.
- Each segment argument uses a format of either
reference:start,stop or reference, where
reference is the ID of the reference object and start and stop are the endpoints of
the region to query, inclusive.
- If the start and stop positions are not provided, they default to the endpoints
of the entire reference sequence. If start and stop are provided at least one
must be a valid position within the reference object, and start may not be
less than 1 or greater than end.
- Multiple segments may be specified.
- type (zero or more)
- Zero or more type IDs to be used for filtering annotations on the type
field. If multiple type names are provided, the resulting list of features
will be the logical OR of the list.
- category (zero or more)
- Zero or more category IDs to be used for filtering annotations by category.
If multiple categories are provided, they are treated as the logical OR.
- categorize (optional)
- Either "yes" or "no" (default). If "yes", then each annotation
must include its functional category.
- feature_id (zero or more)
- Instead of, or in addition to, segment arguments, you may
provide one or more feature_id arguments, whose values
are the identifiers of particular features. If the server
supports this operation, it will translate the feature ID into
the segment(s) that strictly enclose them and return the result
in the features response. It is possible for the server
to return multiple segments if the requested feature is present
on multiple reference objects from different coordinate systems.
- Support for this behaviour must be reported via the feature-by-id
capability.
- group_id (zero or more)
- The group_id argument is similar to feature_id, but
retrieves segments that contain the indicated feature group.
- Support for this behaviour must be reported via the group-by-id
capability.
- maxbins (optional)
- This argument allows a client to indicate to the server the available
rendering space it has for drawing features (i.e. the number of "bins").
The server may choose to alter its response according to this
information, for instance to avoid returning multiple annotations that
would otherwise be rendered in the same pixel and thus be invisible.
It is up to the server to choose how to respond. Support must be indicated
via the maxbins capability.
Note: Although the segment and feature_id
parameters are both optional, you must provide at least one.
Any combination of the two parameters is permitted.
The document returned from the features request is an
XML-formatted "DASGFF" document:
<?xml version="1.0" standalone="no"?>
<DASGFF>
<GFF href="url">
<SEGMENT id="id" start="start" stop="stop" version="X.XX" label="label">
<FEATURE id="id" label="label">
<TYPE id="mytype" category="category" reference="yes|no" cvId="SO:1234">My Type</TYPE>
<METHOD id="mymethod" cvId="ECO:5678">My Method</METHOD>
<START> start </START>
<END> end </END>
<SCORE> [X.XX|-] </SCORE>
<ORIENTATION> [0|-|+] </ORIENTATION>
<PHASE> [0|1|2|-]</PHASE>
<NOTE> note text </NOTE>
<LINK href="url"> link text </LINK>
<TARGET id="id" start="x" stop="y"> target name </TARGET>
<PARENT id="parent id1" />
<PART id="child id1" />
<PART id="child id2" />
</FEATURE>
<FEATURE id="child id1" label="child label">
...
</FEATURE>
<FEATURE id="child id2" label="child label">
...
</FEATURE>
...
<FEATURE id="parent id1" label="parent label">
...
</FEATURE>
...
</SEGMENT>
</GFF>
</DASGFF>
The response XML is formally described by a RELAX NG schema definition, and is
explained below:
- <!DOCTYPE> (required; one only)
- The doctype indicates which formal DTD specification to use.
For the features query, the doctype DTD is "http://www.biodas.org/dtd/dasgff.dtd".
- <DASGFF> (required; one only)
- The appropriate root tag is DASGFF.
- <GFF> (required; one only)
- There is a single <GFF> tag.
- The href (required) attribute
echoes the URL query that was used to fetch the current document.
- The version
attribute indicates the current version of the XML form of the
General Feature Format. The current version is (arbitrarily) 1.0
- <SEGMENT> (required; one or more)
- The <SEGMENT> tag provides information on the reference segment queried.
The id attribute is required.
- The start and stop attributes indicate the position within the segment queried.
The server must include these if it knows them or can deduce them from the request,
but they are otherwise optional.
Note that the start and stop need not necessarily match the request exactly,
for example if the server has more accurate information for the length of a segment.
- The version attribute (optional) indicates
the version of the reference object being annotated, used for coordinate systems which are
not themselves versioned.
- The optional label attribute provides a human readable label for
display purposes. If omitted, it is assumed the ID is appropriate for display.
- The optional type attribute describes the segment type.
- <FEATURE> (optional; zero or more per SEGMENT)
- There are zero or more <FEATURE> tags per <SEGMENT>,
each providing information on one annotation. The
id attribute (required) is an identifier for the feature. it must
be unique to the feature across the data source.
- The label attribute (optional) is a suggested label to
display for the feature. If not present, it is assumed the id attribute is
suitable for display.
- <TYPE> (required; one per FEATURE)
- Each feature has just one <TYPE> field, which indicates the type of the
annotation. The attributes are id (required), which is a unique ID for the
annotation type, category (optional, recommended), which provides functional grouping
to related types, and cvId (optional, recommended)
which is the ID of a term from a relevant controlled vocabulary.
See the Ontology
section for more details.
- The optional reference, subparts and superparts
attributes for the use of reference servers with multiple
coordinate systems in a hierarchy (such as a genomic assembly). They provide
a way to reconstruct the hierarchy, and are described in the Fetching Assembly Information section. Valid values
for these attributes are "yes" and "no". If an attribute is omitted, "no"
is assumed.
- The tag contents (optional) is a human readable label for
display purposes. if Omitted, it is assumed the type ID is appropriate for display.
- Note that it is not permitted for multiple TYPE elements with
the same ID to have different attributes or content to each other.
- The type ID and category may be used
as keys into the stylesheet to select the glyph
and graphical characteristics for the feature.
- <METHOD> (required; one per FEATURE)
- Each feature has one <METHOD> field, which identifies the method used
to identify the feature. The id attribute is required.
- The cvId (optional, recommended) attribute is an
ontology term ID from the Evidence Codes Ontology, and as such is a generic
(potentially less specific) representation of the method.
- The tag contents (optional) is a human readable label representing the name of the method. If omitted, it is
assumed the method ID is suitable for display.
- <START>, <END> (optional; one apiece per FEATURE)
- These tags indicate the start and end of the feature in the coordinate
system of the reference object given in the <SEGMENT> tag. If one
element is present, the other must be also. In addition, start
must not be less than zero and end must not be less than
start.
- If start and end are zero, or omitted entirely, it is assumed that the
feature is an annotation of the reference object as a whole rather than a
region of sequence. These are known as non-positional
annotations.
- <SCORE> (optional; one per FEATURE)
- This is an integer or floating point number indicating the "score" of the method used to find
the current feature. If this field is inapplicable, the contents of the tag can be
replaced with a - symbol. This is the assumed value if the tag is
omitted entirely.
- <ORIENTATION> (optional; one per FEATURE)
- This tag indicates the orientation of the feature relative to the direction of
transcription. It may be 0 for features that are unrelated to transcription,
+, for features that are on the sense strand, and -, for features on the
antisense strand. If this tag is omitted, a value of 0 is assumed.
- <PHASE> (optional; one per FEATURE)
- This tag indicates the position of the feature relative to open reading frame, if
any. It may be one of the integers 0, 1 or
2, corresponding to each of the three reading frames, or - if the
feature is unrelated to a reading frame. If this tag is omitted, a value
of - is assumed.
- <NOTE> (optional; zero or more per FEATURE)
- A human-readable note in plain text format only.
- <LINK> (optional; zero or more per FEATURE)
- A link to a web page somewhere that provides more information about this feature. The
href (required) attribute provides the URL target for the link. The
link text is an optional human readable label for display
purposes.
- <TARGET> (optional; zero or more per FEATURE)
- The target sequence in a sequence similarity match. The id attribute provides the
reference ID for the target sequence, and the start and stop attributes
indicate the segment that matched across the target sequence. All
three attributes are required. The content of the tag (optional) is a human readable label. If omitted,
it is assumed the ID is suitable for display.
- <GROUP> (optional; zero or more per FEATURE)
- The <GROUP> section is slightly odd, as it is derived from an overloaded
field in the GFF flat file format. It provides a unique "group" ID that indicates
when certain features are related to each other. The canonical
example is the CDS, exons and introns of a transcribed gene, which logically
belong together. This element is deleted in favour of the <PARENT> tag.
- The group id attribute (required) provides an identifier that
should be used by the client to group features together visually.
- The label attribute (optional) provides a human-readable string that can be
used in graphical representations to label the glyph.
- The type attribute (optional) provides a type ID for the group as a whole,
for example "transcript". This ID can be used as a key into the
stylesheet to select the glyph and graphical characteristics for the
group as a whole.
- <NOTE> (optional; zero or more per GROUP)
- As documented above.
- <LINK> (optional; zero or more per GROUP)
- As documented above.
- <TARGET> (optional; zero or more per GROUP)
- As documented above. NOTE: although this tag is present in the GROUP section,
it applies to the FEATURE, and it is preferred to place it directly in the <FEATURE>
section. Earlier versions of this specification placed the
TARGET tag in the GROUP section, and clients must recognize and
accomodate this.
- <PARENT>, <PART> (optional; zero or more per FEATURE)
- A replacement for the <GROUP> tag, these tags
identify other features that are parents or children of this feature within a
hierarchy. Each has a single required attribute, id, which refers to
a separate <FEATURE> tag. This mechanism means a parent or child feature
need only be defined once and may be referred to multiple times. This is
preferable to the use of <GROUP> tags, where a parent must be defined
separately for every child. It also allows more than two levels of
hierarchy to be defined (e.g. a gene has parts - transcripts, and transcripts
have parts - exons). In addition, parent features may have start/end positions.
- Note that this approach differs in that a parent-part relationship
must be declared in both features. That is, all parents must include
references to all of their parts using <PART> tags, and all parts
must include references to all of their parents using <PARENT> tags.
Notes
The ID of a feature must be unique across a data source. This
means that no two features may share the same ID. Note that the same feature
annotated onto different reference objects (e.g. an exon annotated onto a
contig and chromosome) do not need different IDs.
Annotation servers are required to return all annotations which overlap the
indicated segment, and not just those that are completely contained within it.
In addition, if any overlapping features have parents or parts, all features
within the parent/part hierarchy must also be provided in the
response, regardless of whether they overlap the query segment.
For example:
Query +=================+
| X |
| |
| |
-------+-----------------+------------------
| A |
| | -----------
Features | | B
| |
------ | ---- ------ -+---------- ----
A1 | A2 A3 | A4 A5
| |
| | ---- --
| | B1 B2
The above diagram shows the locations of nine annotations relative to a query
range X. The features are organised into two parent/part hierarchies,
A and B. Annotations A1 to A5 are parts of annotation A, and features B1 to B2 are
parts of annotation B.
For this query the server will return the parent annotation A because it
spans the query range X entirely, the child annotations A2 and A3
because they lie wholly within the query range, child annotation A4
because it overlaps partially with X, and child annotations A1 and A5 because
they are within the same parent/part hierarchy as at least one of these
annotations. Annotations B, B1 and B2 should not be returned as none of them
overlap X.
Annotations must be returned using the
coordinate system in which they were requested. For example, if a contig ID
was used to specify the segment, then the annotation endpoints must use contig coordinates.
If multiple segment arguments are provided and they happen to overlap,
then the annotation server may return the same annotation multiple
times, possibly using different coordinate systems. It is the
responsibility of the client to merge annotations based on the assembly. Note
that the ID of duplicate features will be the same, thus allowing them to be
filtered.
Description:
This query can be issued to an annotation server in order to retrieve
the server's recommendations on formatting annotations retrieved from
it. These recommendations are not normative. A viewer is free to use
any display format it chooses.
Scope:
Annotation servers.
Request:
This command is executed relative to a data source:
SERVER/das/DSN/stylesheet
Arguments:
None.
Response:
The response to the stylesheet command is the "DASSTYLE" XML-formatted
document:
<?xml version="1.0" standalone="no"?>
<DASSTYLE>
<STYLESHEET version="X.XX">
<CATEGORY id="default">
<TYPE id="default">
<GLYPH zoom="high">
<ID>
<ATTR>value</ATTR>
<ATTR>value</ATTR>
...
</ID>
</GLYPH>
<GLYPH zoom="medium">
<ID>
<ATTR>value</ATTR>
<ATTR>value</ATTR>
...
</ID>
</GLYPH>
<GLYPH zoom="low">
<ID>
<ATTR>value</ATTR>
<ATTR>value</ATTR>
...
</ID>
</GLYPH>
</TYPE>
</CATEGORY>
<CATEGORY id="group">
<TYPE id="group_id1">
<GLYPH zoom="high">
<ID>
<ATTR>value</ATTR>
<ATTR>value</ATTR>
...
</ID>
</GLYPH>
...
</TYPE>
</CATEGORY>
<CATEGORY id="category1">
<TYPE id="default">
<GLYPH>
<ID>
<ATTR>value</ATTR>
...
</ID>
</GLYPH>
</TYPE>
<TYPE id="type1">
<GLYPH>
<ID>
<ATTR>value</ATTR>
...
</ID>
</GLYPH>
</TYPE>
<TYPE id="type2">
<GLYPH>
<ID>
<ATTR>value</ATTR>
...
</ID>
</GLYPH>
</TYPE>
...
</CATEGORY>
<CATEGORY id="category2">
<TYPE id="default">
<GLYPH>
<ID>
<ATTR>value</ATTR>
...
</ID>
</GLYPH>
</TYPE>
...
</CATEGORY>
...
</STYLESHEET>
</DASSTYLE>
This document is intended to provide hints to the annotation display
client. It maps feature categories and individual types to a series
of glyphs known to the display client.
The response XML is formally described by a RELAX NG schema definition, and is
explained below:
- <!DOCTYPE> (required; one only)
- The doctype indicates which formal DTD specification to use.
For the stylesheet query, the doctype DTD is "http://www.biodas.org/dtd/dasstyle.dtd".
- <DASSTYLE> (required; one only)
- The appropriate root tag is DASSTYLE.
- <STYLESHEET> (required; one only)
- There is a single <STYLESHEET> tag.
- The version
(required) attribute indicates the current version of the stylesheet, and
can be used for caching purposes.
- <CATEGORY> (required; one or more)
- There are one or more <CATEGORY> tags, each providing information
on the display of a high-level feature category. The id
(required) atribute uniquely names the category.
- A special category name is "default", which
tells the annotation viewer what format to use for categories that
are not otherwise specified in the stylesheet.
- Another special category name is "group". A "group" entry indicates the
format to use for groups of features.
- <TYPE> (required; one or more per CATEGORY)
- There are one or more <TYPE> tags per <CATEGORY>,
each providing display suggestions for one type of annotation.
The id (required) attribute uniquely identifies the type.
- Where enclosed in a category tag with the special "group" ID, a type tag
correspond to the type of a feature's group, rather than the feature.
- A special type ID is "default", which
tells the annotation viewer what format to use for feature types in the
enclosing category that are not otherwise specified in the stylesheet.
- <GLYPH> (required; one or more per TYPE)
- There is one or more <GLYPH> tag per <TYPE>. It contains within it
information on what glyph (graphical widget) to use to display
the indicated annotation type.
- The optional zoom attribute,
implements a simple form of semantic zooming, and allows the client
to select the glyph and its attributes based on the zoom level. Possible
values are "high", "medium" and "low". If multiple <GLYPH> tags
are present, this attribute must be present in order to select
among them. A "high" zoom means that there are fewer base pairs per
pixel (high magnification). A "low" zoom shows more base pairs. "Medium" is
intermediate. It is left to the client to
determine the boundaries for "high", "medium" and "low", since this is a
function of the graphics rendering.
- <ID> (required; one per GLYPH)
- The value of ID is one of the recognised glyph types.
- <ATTR> (optional; one or more per ID)
- Each glyph type may have one or more optional elements within it to
determine rendering properties (e.g. colour). The value of ATTR
therefore depends on the glyph type.
Here is a short stylesheet example:
<?xml version="1.0" standalone="no"?>
<DASSTYLE>
<STYLESHEET version="1.0">
<CATEGORY id="similarity">
<TYPE id="default">
<GLYPH>
<LINE>
<FGCOLOR>gray</FGCOLOR>
</LINE>
</GLYPH>
</TYPE>
<TYPE id="NN">
<GLYPH >
<BOX>
<HEIGHT>4</HEIGHT>
<FGCOLOR>black</FGCOLOR>
<BGCOLOR>red</BGCOLOR>
</BOX>
</GLYPH>
</TYPE>
<TYPE id="NP">
<GLYPH>
<TOOMANY>
<HEIGHT>4</HEIGHT>
<FGCOLOR>black</FGCOLOR>
<BGCOLOR>blue</BGCOLOR>
</TOOMANY>
</GLYPH>
</TYPE>
<TYPE id="PN">
<GLYPH>
<BOX>
<HEIGHT>3</HEIGHT>
<FGCOLOR>blue</FGCOLOR>
<BGCOLOR>green</BGCOLOR>
</BOX>
</GLYPH>
</TYPE>
<TYPE id="PP">
<GLYPH>
<SPAN>
<HEIGHT>4</HEIGHT>
<FGCOLOR>gray</FGCOLOR>
</SPAN>
</GLYPH>
</TYPE>
</CATEGORY>
</STYLESHEET>
</DASSTYLE>
Glyphs and Groups
Glyphs and their attributes are typically applied to individual
features. However, they can be applied to entire groups as well (via
the type attribute of the <GROUP> tag in the features command).
In this case, the glyph will apply to the connecting regions between
the features of the group.
For example, to indicate that the exons in a "transcript" group should
be drawn with a yellow box, that the UTRs should be drawn with a blue
box, and that the connections between exons should be drawn with a
hat-shaped line:
<CATEGORY id="transcription">
<TYPE id="exon">
<GLYPH>
<BOX>
<BGCOLOR>yellow</BGCOLOR>
</BOX>
</GLYPH>
</TYPE>
<TYPE id="utr">
<GLYPH>
<BOX>
<BGCOLOR>blue</BGCOLOR>
</BOX>
</GLYPH>
</TYPE>
</CATEGORY>
<CATEGORY id="group">
<TYPE id="transcript">
<GLYPH>
<LINE>
<FGCOLOR>black</FGCOLOR>
<STYLE>hat</STYLE>
</LINE>
</GLYPH>
</TYPE>
...
Note that <GROUP> tags are deprecated in favour of linking to explicit
features via the <PARENT> and <PART> tags. When these are used,
styles for parent features should be defined via the standard category/type
mechanism.
Description:
This query returns a protein 3D structure, including metadata and coordinates.
Scope:
Reference servers.
Request:
This command is executed relative to a data source:
SERVER/das/DSN/structure?query=STRUCTUREID
[;chain=CHAINID ...]
[;model=MODELNUM ...]
Arguments:
- query (required)
- This is the ID of the reference structure.
- chain (optional; zero or more)
- Limits the response to only contain the chain with the given ID. If
omitted, all available chains are returned.
- model (optional; zero or more)
- If the query structure has been resolved using NMR, several alternate
models are available. Using this argument it is possible to request only
individual models. If omitted, all available models are returned.
Arguments of different types (e.g. query and chain) are interpreted as logical
intersections. Arguments of the same type (e.g. chain=A and chain=B) are
interpreted as logical unions. Thus a query might look like:
"get structures where the query ID is 2ii9 AND the chain is A OR B".
Response:
The document returned from the structure request is an
XML-formatted "DASSTRUCTURE" document:
<?xml version="1.0" standalone="no"?>
<dasstructure>
<object dbAccessionId="someid" objectVersion="version" dbSource="someDB" dbVersion="version" dbCoordSys="coords"/>
<objectDetail dbSource="someDB" property="property">
some details about the object. e.g. description, etc.
</objectDetail>
<chain id="chainID" model="modelNum" SwissprotId="accessionCode">
<group name="groupName" type="groupType" groupID="groupID" insertCode="iCode">
<atom x="xCoord" y="yCoord" z="zCoord" atomName="atomname" atomID="atomID" occupancy="occupancy" tempFactor="tempFactor" altLoc="altLoc"/>
</group>
</chain>
<connect type="connectionType" atomSerial="atomID" >
<atomID atomID="atomID"/>
</connect>
</dasstructure>
The response XML is formally described by a RELAX NG schema definition, and is
explained below:
- <dasstructure> (required; one only)
- The appropriate root tag is dasstructure.
- <object> (required; one or more)
- Provides basic details of each structure object.
- The dbAccessionId (required) attribute is a unique identifier for
the structure record - that is, the query ID.
- The objectVersion
(required) attribute identifies the version of the structure. The content
of the attribute depends on the coordinate system. See the
Reference and Annotation Servers section for more details.
- The dbSource (required) attribute is the name of the database
containing the structure.
- The dbVersion (required) is the version of the database containing
the structure.
- The dbCoordSys (required) attribute is the text description of the
object's coordinate system, e.g. PDBresnum,Protein Structure.
- <objectDetail> (optional; zero or more)
- Provides additional key-value details of each structure object. The
property (required) attribute identifies the name of the property,
and the dbSource (required) attribute identifies the source of the
property. The tag contents (required) provide a free-text value for the
property.
- <chain> (optional; zero or more)
- Represents a single structural chain.
The id (required) attribute is the identifier for the chain, e.g.
"A" or "B". The model (optional) attribute is the model number of
the chain, where applicable (e.g NMR structures).
- <group> (optional; zero or more)
- Each chain has zero or more group elements, each representing a
group of atoms such as an amino acid or a hetero molecule.
- The type (required) attribute describes the type of group. It may
be one of: amino, nucleotide, hetatom.
- The name (required) attribute is the name of the group, e.g. "ALA"
and groupID (required) is a unique identifier within the structure.
- The insertCode (optional) attribute is an uppercase alphabet
character (A-Z) used to distinguish sequential groups with the same
residue number. For example amino acid "86A" might be the 87th group in a
chain, with an insertCode of "A".
- <atom> (required; one or more)
- Each group has one or more atom elements, each representing a
single atom in a single conformation.
- The x, y and z (required) attributes are floating
point numbers describing the coordinates of the atom. The atomID
(required) attribute uniquely identifies the atom within the structure
and the atomName (required) attribute is a name (symbol) for the
atom.
- The occupancy (optional) and tempFactor (optional)
attributes are floating point numbers representing the occupancy and
temperature factor of the atom, respectively.
- The altLoc (optional) attribute indicates that this definition
describes one of several possible locations for the atom. Different atoms
with the same altLoc consitute a single conformation.
- For full details of these
attributes, see the wwPDB documentation.
- <connect> (optional; zero or more)
- Each dasstructure may have zero or more connect elements,
each representing an inter-atom connection. Amino acid connections do not
need to be included, but for hetatoms this is mandatory (as otherwise a
viewer would not know how to display them).
- The type (required) attribute describes the type of connection
(e.g. bond), and the atomSerial (required) attribute is
the atomID of the source of the bond.
- <atomID> (required; one or more)
- Each target atom within the connection is represented by an atomID
element. The element has a single atomID (required) attribute,
which is the atomID of the target atom.
Example
<?xml version='1.0' standalone='no' ?>
<dasstructure>
<object dbAccessionId="2ii9"
objectVersion="20-MAR-07"
dbSource="PDB"
dbVersion="20070116"
dbCoordSys="PDBresnum,Protein Structure" />
<chain id="A">
<group name="SER" type="amino" groupID="1">
<atom atomID="1" atomName="N" x="44.18" y="5.327" z="31.168" />
<atom atomID="2" atomName="CA" x="43.672" y="5.068" z="29.781" />
<atom atomID="3" atomName="C" x="42.728" y="6.217" z="29.365" />
<atom atomID="4" atomName="O" x="42.328" y="7.024" z="30.23" />
<atom atomID="5" atomName="CB" x="42.965" y="3.707" z="29.74" />
<atom atomID="6" atomName="OG" x="42.754" y="3.284" z="28.41" />
</group>
...
</chain>
...
</dasstructure>
dsn [deprecated]
Description:
This query returns the list of data sources that are available from this server.
Scope:
Reference and annotation servers.
Request:
This command is executed relative to the server:
SERVER/das/dsn
Response:
The response to the dsn command is the "DASDSN" XML-formatted document:
<?xml version="1.0" standalone="no"?>
<!DOCTYPE DASDSN SYSTEM "http://www.biodas.org/dtd/dasdsn.dtd">
<DASDSN>
<DSN>
<SOURCE id="id1" version="version">source name 1</SOURCE>
<MAPMASTER>URL</MAPMASTER>
<DESCRIPTION>descriptive text 1</DESCRIPTION>
</DSN>
<DSN>
<SOURCE id="id2" version="version">source name 2</SOURCE>
<MAPMASTER>URL</MAPMASTER>
<DESCRIPTION href="url">descriptive text 2</DESCRIPTION>
</DSN>
...
</DASDSN>
- <!DOCTYPE> (required; one only)
- The doctype indicates which formal DTD specification to use.
For the dsn query, the doctype DTD is "http://www.biodas.org/dtd/dasdsn.dtd".
- <DASDSN> (required; one only)
- The appropriate root tag is DASDSN.
- <DSN> (required; one or more)
- There are one or more <DSN> tags, one for each data
source. Each <DSN> contains one <SOURCE> tag,
one <MAPMASTER> tag, and
optionally one <DESCRIPTION> tag.
- <SOURCE> (required; one per DSN tag)
- This tag indicates the symbolic name for a data source. The
symbolic name to use for further requests can be found in the
id (required) attribute. A source version
attribute is optional, but strongly recommended.
The tag body contains a human-readable
label which may or may not be different from the ID.
- <MAPMASTER> (required; one per DSN tag)
- This tag contains the URL (SERVER/das/DSN) of the reference server for
the coordinate system annotated by this data source. For an annotation
server, this will be a different server. For a reference server, this
would echo its own URL.
- <DESCRIPTION> (optional)
- This tag contains additional descriptive information about the
data source. If an href (optional) attribute is present, the
attribute contains a link to further human-readable information
about the data source, such as its home page.
dna [deprecated]
Description:
This query returns the DNA sequence corresponding to the indicated segment.
Scope:
Reference servers
Request:
This command is executed relative to a data source:
SERVER/das/DSN/dna?segment=RANGE[;segment=RANGE...]
Arguments:
- segment (required; one or more)
- Each segment argument uses a format of either
reference:start,stop or reference, where
reference is the ID of the reference object and start and stop are the endpoints of
the region to query, inclusive.
- If the start and stop positions are not provided, they default to the endpoints
of the entire reference sequence. If start and stop are provided at least one
must be a valid position within the reference object, and start may not be
less than 1 or greater than end.
Here is an example of a valid request that uses the segment
argument to fetch three non-overlapping segments:
http://www.ensembl.org/das/Homo_sapiens.NCBI36.reference/dna?segment=Y;segment=X:1,1000;segment=1:50,200
Response:
The response to dna is the "DASDNA" XML-formatted document.
<?xml version="1.0" standalone="no"?>
<!DOCTYPE DASDNA SYSTEM "http://www.biodas.org/dtd/dasdna.dtd">
<DASDNA>
<SEQUENCE id="id" start="start" stop="stop" version="X.XX">
<DNA length="NNNN">
atttcttggcgtaaataagagtctcaatgagactctcagaagaaaattgataaatattat
taatgatataataataatcttgttgatccgttctatctccagacgattttcctagtctcc
agtcgattttgcgctgaaaatgggatatttaatggaattgtttttgtttttattaataaa
taggaataaatttacgaaaatcacaaaattttcaataaaaaacaccaaaaaaaagagaaa
aaatgagaaaaatcgacgaaaatcggtataaaatcaaataaaaatagaaggaaaatattc
agctcgtaaacccacacgtgcggcacggtttcgtgggcggggcgtctctgccgggaaaat
tttgcgtttaaaaactcacatataggcatccaatggattttcggattttaaaaattaata
taaaatcagggaaatttttttaaattttttcacatcgatattcggtatcaggggcaaaat
tagagtcagaaacatatatttccccacaaactctactccccctttaaacaaagcaaagag
cgatactcattgcctgtagcctctatattatgccttatgggaatgcatttgattgtttcc
gcatattgtttacaaccatttatacaacatgtgacgtagacgcactgggcggttgtaaaa
cctgacagaaagaattggtcccgtcatctactttctgattttttggaaaatatgtacaat
gtcgtccagtattctattccttctcggcgatttggccaagttattcaaacacgtataaat
aaaaatcaataaagctaggaaaatattttcagccatcacaaagtttcgtcagccttgtta
tgtcaaccactttttatacaaattatataaccagaaatactattaaataagtatttgtat
gaaacaatgaacactattataacattttcagaaaatgtagtatttaagcgaaggtagtgc
acatcaaggccgtcaaacggaaaaatttttgcaagaatca
</DNA>
</SEQUENCE>
</DASDNA>
- <!DOCTYPE> (required; one only)
- The doctype indicates which formal DTD specification to use.
For the dna query, the doctype DTD is "http://www.biodas.org/dtd/dasdna.dtd".
- <DASDNA> (required; one only)
- The appropriate doctype and root tag is DASDNA.
- <SEQUENCE> (required; one or more)
- There is a single <SEQUENCES> tag per requested segment. It has the
attributes id, which indicates the reference ID for this sequence,
start and stop, which indicate the position of
this segment within the reference sequence, and version,
which provides the sequence map version number. All four
attributes are required.
- <DNA> (required; one per SEQUENCE)
- This tag surrounds the DNA data. It has the attribute
length (required), which indicates the length of the DNA.
The DNA is found in the body of the tag and is required.
DNA will be lower-case and adhere to the IUPAC code conventions.
link [deprecated]
Description:
This query can be issued in order to retrieve further human-readable
information about an annotation. It is best to pass this URL directly to a
browser, as the type of the returned data is not specified (it will typically
be an HTML file, but any MIME format is allowed). This command is
deprecated due to lack of use.
Scope:
Annotation servers.
Request:
This command is executed relative to a data source:
SERVER/das/DSN/link?field=TAG;id=ID
Arguments:
- field (required)
- The field to fetch further information on. Options are:
- feature -- the feature itself
- type -- the feature type
- method -- the feature method
- category -- the feature category
- target -- the target, applicable to sequence
similarities only
- id (required)
- The ID of the indicated annotation field.
Response:
The document returned from the link request may be any
browser-readable MIME format.
This section describes the procedure a server should use for handling requests
in the sequence, features and types commands
from a client where one or more of the requested identifiers are in some way
invalid. In such cases it replaces the system of using HTTP status codes since it
allows for requests which contain both valid and invalid identifiers.
Support for this functionality is reported by the unknown-segment,
error-segment and unknown-feature
capabilities. Annotation servers should report
unknown-segment and unknown-feature, and reference servers should indicate
error-segment instead of unknown-segment.
Note that an annotation server may be unable to support the unknown-segment
capability (e.g. for performance reasons). In such cases, the client will be
unable to distinguish between a lack of annotations in the specific region
requested and a lack of annotations across the whole reference object. Both will
appear as empty <SEGMENT> elements.
Note that a reference server is required to support the
error-segment capability, complementing the mandatory implementation
of the entry_points command.
A request for sequence, features or types may fail
because:
- the requested segment is an invalid format (e.g. start is present but end is missing, start is less than 1, or start is greater than end)
- the requested segment is wholly outside the bounds of the reference object
- the reference object or feature is not known to the server
In these cases, an exception is indicated by issuing an <ERRORSEGMENT>,
<UNKOWNSEGMENT> or <UNKOWNFEATURE> tag instead of the usual
<SEGMENT> tag. The tag has an id attribute (required)
corresponding to the ID of the requested segment or feature. <ERRORSEGMENT>
and <UNKNOWNSEGMENT> elements may also have start and
stop (optional) attributes corresponding to the requested bounds of the
segment (if this was specified).
In the case of a request for multiple segments/features, the server will return a
mixture of <SEGMENT> sections for valid segments, and
exception elements for invalid ones:
<ERRORSEGMENT id="id" start="start" stop="stop" />
<UNKNOWNFEATURE id="id" />
<SEGMENT id="id" start="start" stop="stop" version="version">
...
</SEGMENT>
A server will raise different types of exception in different circumstances.
This is best illustrated via a flow diagram:

To explain, a reference server knows that any reference object it cannot identify must be
erroneous. It will therefore always raise <ERRORSEGMENT>s. By contrast
an annotation server, which is not required to know the identities of all the
reference objects in the coordinate system, will typically respond by issuing
an <UNKNOWNSEGMENT> tag when it does not recognise a reference object -
it does not know whether the request is erroneous or not. Note that all
servers should issue <ERRORSEGMENT> exceptions when they detect a query
segment that is invalid or wholly outside the range of a reference object.
Reference servers for hierarchical coordinate systems such as genomic assemblies
must provide a mechanism for reconstructing the relationships between the
reference objects from different coordinate systems in the hierarchy. This is
accomplished using the Features command in a specific
manner.
The client requests a list of features representing the reference objects
either below or above a query segment in the hierarchy. It does this by
specifying a category parameter of either "component" or "supercomponent",
respectively.
The server returns features representing these reference objects with
a category attribute of either "component" or "supercomponent" accordingly.
Each
also has a reference attribute of "yes" to indicate that the feature
represents a reference object and therefore is itself an entry point.
If a reference object within the assembly contains other reference objects
which are themselves reference objects, the feature will also have a
subparts attribute of "yes". Likewise, reference objects which have
other objects above them in the hierarchy will have a superparts
attribute of "yes". Components that are the
parents of the reference sequence in the assembly have a category
attribute of "supercomponent".
Moving Down in an Assembly
For those components that have subparts, the start and end of the
feature give the feature's position in the requested segment's
coordinate system, and the id, start and end of the <TARGET>
element gives the feature's position in its native coordinates.
For example:
1 200 400 1000
+--------+-----------+-------------------+ 22
1 200 220 1 20 620
+--------+---- A --+-------------------+ B
1 80 280 400
------+-----------+-------- C
=================== C.1
============= C.2
A request for this assembly will look like the following:
http://www.ensembl.org/das/Homo_sapiens.NCBI36.reference/features?segment=22:1,1000;category=component
The reference server will return the following (abbreviated) document:
<SEGMENT id="22" start="1" stop="1000">
<FEATURE id="22">
<START>1</START>
<END>1000</END>
<TYPE id="Chromosome" category="component" reference="yes" superparts="no" subparts="yes">Chromosome</TYPE>
<TARGET id="22" start="1" stop="1000">22</TARGET>
...
</FEATURE>
<FEATURE id="Contig-A">
<START>1</START>
<END>200</END>
<TYPE id="Contig" category="component" reference="yes" superparts="yes" subparts="no">Contig</TYPE>
<TARGET id="Contig-A" start="1" stop="200">Contig A</TARGET>
...
</FEATURE>
<FEATURE id="Contig-B">
<START>400</START>
<END>1000</END>
<TYPE id="Contig" category="component" reference="yes" superparts="yes" subparts="no">Contig</TYPE>
<TARGET id="Contig-B" start="20" stop="620">Contig B</TARGET>
...
</FEATURE>
<FEATURE id="Contig-C">
<START>200</START>
<END>400</END>
<TYPE id="Contig" category="component" reference="yes" superparts="yes" subparts="yes">Contig</TYPE>
<TARGET id="Contig-C" start="80" stop="280">Contig C</TARGET> ...
</FEATURE>
</SEGMENT>
Notice that contig C is marked as having subparts. This is an
indication to the client that it should emit a features request that
includes segment C:80,280 in order to discover its components (C.1 and
C.2).
Notice also that chromosome 22 appears as a component of itself with the
attribute superparts="no" and subparts="yes". This is a
side effect of providing information about the component parent.
Moving Up in an Assembly
It is also desirable for a client to fetch the parent of a
segment, so as to accomodate the situation in which the user enters
the browser at a contig or sequenced clone, and wants to "zoom out."
This situation is complicated by rough draft issues, in which a single
rough draft sequence segment may have multiple parents, and some
sections of the segment may not belong in the assembly at all. For
example:
A B C D
contig21-----------> <-----------contig100
| | / /
| | / /
Acc A ---------------------
a b c d
Here, the segment "Acc A" contains two fragments, one of which is
located on contig21 and the other on contig100.
To retrieve this information, the client requests the category
supercomponent. For segments that are in the middle of the
assembly, one or more assembly parents will be returned in
addition to subcomponents. The parent <START>,
<END> and <ORIENTATION> tags are presented in the
coordinate system of the requested segment, as always. The
start and stop attributes of the <TARGET> tag,
denote the corresponding segment in the coordinate system of the
parent. As always, start is less than stop, for both the feature and
the target.
<SEGMENT id="Acc A" start="1" stop="1000">
<FEATURE id="contig21">
<START>a</START>
<END>b</END>
<ORIENTATION>+</ORIENTATION>
<TYPE id="Contig" category="supercomponent" reference="yes" superparts="yes" subparts="yes">a contig</TYPE>
<TARGET id="contig21" start="A" stop="B"></TARGET>
</FEATURE>
<FEATURE id="contig100">
<START>c</START>
<END>d</END>
<ORIENTATION>-</ORIENTATION>
<TYPE id="Contig" category="supercomponent" reference="yes" superparts="yes" subparts="yes">a contig</TYPE>
<TARGET id="contig100" start="C" stop="D"></TARGET>
</FEATURE>
</SEGMENT>
To continue following the parents upward in the assembly, the client
will issue further features requests for the target IDs, in this case
"contig21" and "contig100". In the general case, following parents
will project the requested segment onto a discontinuous set of
regions, potentially on different chromosomes. The client may wish to
alert the user and refuse to proceed further when it encounters a
segment with multiple parents.
Annotations returned by the features command have a single type
tag. Each type has an ID, used to identify features of the same type within
the DAS source, a human readable description of the type intended for display,
an ontology term ID and a category. This section describes the content of
these fields.
Often, different DAS annotation sources provide similar features. It is useful
for client software to have a way to formally categorise these annotations so
that they can be presented to the user in an intelligent manner. This also
allows a user to more easily access the types of data they are interested in,
which is especially important given the now large number of DAS servers. To facilitate
this, the protocol now incorporates formal ontologies. Previous
versions of the DAS specification had only a rudimentary classification of
feature types (see below), and this did not extend to protein features.
The TYPE and METHOD elements in the Features
and Types commands have a cvId attribute
(meaning "controlled vocabulary identifier"). This can be
populated with the ID of a term from one of a selection of ontologies, which
a client can then choose to use with its own representation. Though
these attributes are optional, it is highly recommended they be included.
Use of the ontology allows for much greater visibility of the data, both in
clients and in the DAS Registry, which allows users to search by ontology term
to find all servers with similar types of annotations. In addition,
these attributes may be required in future versions of the protocol.
TYPE elements may be annotated with a cvId from the following ontologies:
-
Sequence Types and Features (namespace: SO) - contains terms for both
genomic and protein sequence annotations.
-
Protein Modifications (namespace: MOD) - contains terms for post-translational
modifications.
-
BioSapiens Annotations (namespace: BS) - originally developed for the BioSapiens
consortium, this ontology contains protein-focussed terms for nonpositional
annotations (e.g. publications).
METHOD elements may be annotated with a cvId from the following ontologies:
-
Evidence Codes (namespace: ECO) - contains terms for describing the source
of data (e.g. author statements, direct assays, computational analysis).
Note: The BS and SO ontologies overlap due
to their respective scopes, but equivalent terms are aliased appropriately.
DAS client developers may choose to implement any ontology term processing
functionality they posess (such as sorting, filtering or reasoning) with the
ability to identify these links.
Note: In future versions of the specification it is
anticipated that the number of permitted ontologies will be increased.
Choosing Terms
Through the use of these ontologies, an annotation from any one source may be
"genericised" such that it may be directly compared with annotations from
other sources. To this end, data providers should select ontology terms that
are as specific as possible, but should never include reference to the source
of the annotation (e.g. "Pfam domain"). Instead, the term is intended to
describe only the class of data. More specific unstructured
information may be provided in the CDATA of the appropriate element.
For example, for a protein domain derived automatically using a
specific algorithm, the appropriate term to describe the type of data might be
"polypeptide_domain", and the term to describe the method might be "inferred
from electronic annotation":
<TYPE id="my_domain" cvId="SO:0000417" >My Domain</TYPE> <!-- polypeptide_domain -->
<METHOD id="my_method" cvId="ECO:00000067">My Algorithm</METHOD> <!--inferred from electronic annotation -->
The Ontology Lookup Service
provides a human interface to aid data providers in selecting terms, and also
provides a web service interface which clients may wish to use in reconstructing
the relationships between terms.
No Suitable Term?
Terms may only be selected from one of the ontologies listed above. This is
in order to eliminate redundancy of terms and maintain relationships between
types. Though the ontologies are relatively well established, some features
may not be satisfactorily represented with existing terms. In these cases, a
data provider should request a suitable term be added by contacting the
administrators. If in doubt, consult the DAS mailing list.
This process has proved efficient and productive in the past -
for example, the Sequence Types and Features ontology originally focussed on
genomic sequence features, but now encompasses protein sequences too.
Whereas the ontology term ID is constrained and is thus useful for programatic
processing of annotations, the type ID and type category are not. However,
some categories have special meanings as described in the
Fetching Sequence Assemblies section. In addition,
genomic annotations have historically been categorised according to a short
list of possible categories.
component
This special category indicates that the feature is a child component
of the reference sequence in the current assembly. When combined with the
reference="yes" attribute, this indicates that the feature can
be used as a reference point to retrieve subfeatures contained within
it (including subcomponents).
supercomponent
This special category indicates that the feature is the parent of the
reference sequence in the current assembly. When combined with the
reference="yes" attribute, this indicates that the feature can
be used as a reference point to retrieve features that completely
contain the selected range of the reference sequence.
translation
The translation category is used for features that relate
to regions of the sequence that are translated into proteins.
Features that relate to transcription are separate (see below).
Feature types:
- stop - position of the translation stop codon
- ATG - position of the start codon
- CDS - position of the coding region
- 5'UTR - untranslated region
- 3'UTR - untranslated region
- misc_translated - miscellaneous
transcription
The transcription category is used for features that relate
to regions of the sequence that are transcribed into RNA.
Feature types:
- exon
- intron
- tRNA
- mRNA
- ncRNA
- 5'Cap - transcriptional start site
- PolyA
- Splice5 - splice donor
- Splice3 - splice acceptor
- misc_transcribed
variation
The variation category is used for features that relate
to regions of the sequence that are polymorphic.
Feature types:
- insertion
- deletion
- substitution
- misc_variation
structural
The structural category is used for features that relate
to mapping, sequencing and assembly, as well as for various landmarks
that carry no intrinsic biological information.
Feature types:
- clone
- primer_left
- primer_right
- oligo
- assembly_tag
- misc_structural
similarity
The similarity category is used for areas that are
similar to other sequences. Similarity features should have a
<METHOD> tag that indicates the algorithm used for the sequence
comparison, and a <TARGET> tag that indicates the target of the
match.
Feature types:
- NN -- nucleotide to nucleotide similarity (e.g. blastn)
- NP -- nucleotide to protein similarity (e.g. blastx)
- PN -- protein to nucleotide similarity (e.g. tblastn)
- PP -- protein to protein similarity (e.g. tblastx)
- misc_homology
repeat
The repeat category is used for areas that contain
repetitive DNA. This category is used both for low-complexity
regions, such as microsatellites, and for more biologically
interesting features, such as transposon insertion sites.
Feature types:
- microsatellite
- inverted
- tandem
- transposable_element
- LINE - long repeat not definitely identified as a transposon
- misc_repeat
experimental
The experimental category is a catchall used to flag
areas where there is interesting experimental data of one sort or
another. It is intended for use with high-throughput functional
genomics work, such as knockouts or insertional mutagenesis screens.
Feature types:
- knockout
- expression_tag
- microarrayed
- RNAi_result
- transgenic
- mutant - a mutant phenotype associated with region
- misc_experimental
This section describes a set of generic "glyphs" that can be used by
DAS display programs to display the position of features relative to a
reference object. The annotation server may use these glyphs to send
display suggestions to the viewer via the stylesheet document.
In the descriptions of the glyphs
below, all references to direction are intended to be relative to the plane of
the reference object. For example, when annotating sequences, "width" generally
refers to a dimension in the same plane as (i.e. parallel to) the reference
sequence, whereas "height" is orthogonal:

The current set of glyph ID values are:
- ARROW
- ANCHORED_ARROW
- BOX
- CROSS
- DOT
- EX
- HIDDEN
- LINE
- SPAN
- TEXT
- TOOMANY
- TRIANGLE
- PRIMERS
- GRADIENT
- HISTOGRAM
- LINEPLOT
Glyph Attributes
Each glyph has a set of attributes associated with it which are used to
allow the DAS server indicate the size, color and other visual properties of a
glyph. Attributes are typically optional, but unless otherwise stated the
default for each is determined by the client and may change over time. Thus
DAS servers should set important values even if the default value for any one
client appears to be acceptable. For example if a feature should always be
accompanied by a label, the appropriate attribute should be explicitly set to
"yes".
Attribute values come in the following flavors:
- INT
- An integer
- FLOAT
- A floating point number
- STRING
- A text string
- COLOR
- A color. Colors can be specified using the "#RRGGBB" format
commonly used in HTML, or as one of the
X11 Color set
names recognized by web browsers.
- BOOL
- A boolean value, either "yes" or "no".
- FONT
- A font. Any cross-platform font identifier recognized by Web browsers
is acceptable, e.g. "helvetica".
- ENUM
- One of a pre-defined selection of values, depending on the attribute.
ARROW
A double-headed arrowed line with an axis either orthogonal or parallel to
the sequence map. The direction of the arrow is statically defined, and is not
dependent on the feature's orientation. The arrow covers the extent of the feature.
Attributes
| Attribute | Type | description |
|---|
| PARALLEL |
BOOL |
Arrows run either parallel ("yes") or orthogonal ("no") to the
reference object's axis. |
| SOUTHWEST |
BOOL |
Whether to draw the arrowhead that points west/south (for
parallel/orthogonal arrows). Either "yes" or "no". |
| NORTHEAST |
BOOL |
Whether to draw the arrowhead that points east/north (for
parallel/orthogonal arrows). Either "yes" or "no". |
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The color of the arrow (stroke). |
| BGCOLOR |
COLOR |
The fill color of the area behind the arrow. If omitted, a transparent background is assumed. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Examples
This example shows an arrow with both arrowheads, drawn parallel to the
sequence map:
<ARROW>
<HEIGHT>10</HEIGHT>
<FGCOLOR>purple</FGCOLOR>
<PARALLEL>yes</PARALLEL>
<NORTHEAST>yes</NORTHEAST>
<SOUTHWEST>yes</SOUTHWEST>
<LABEL>yes</LABEL>
<BUMP>yes</BUMP>
</ARROW>

As above, but with <NORTHEAST>no</NORTHEAST>, which
disables the east-facing arrowhead:

An arrow with both arrowheads, drawn perpendicular to the sequence map:

As above but with <NORTHEAST>no</NORTHEAST>, which
disables the north-facing arrowhead:

ANCHORED_ARROW
An arrowed line that has an arrowhead at one end, and an "anchor" (typically
a diamond or orthogonal line) at the other. Unlike ARROW glyphs,
an ANCHORED_ARROW points in the direction indicated by the feature's
<ORIENTATION> tag. It is sized to cover the
extent of the feature. The directionality of the arrow for features with no
orientation is not defined, and therefore this glyph is not recommended for
such features.
Attributes
| Attribute | Type | description |
|---|
| PARALLEL |
BOOL |
Arrows run either parallel ("yes") or orthogonal("no") to the
reference object's axis. |
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The color of the arrow (stroke). |
| BGCOLOR |
COLOR |
The fill color of the area behind the arrow. If omitted, a transparent background is assumed. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Examples
This example shows an anchored arrow drawn parallel to the sequence map:
<ANCHORED_ARROW>
<HEIGHT>10</HEIGHT>
<FGCOLOR>darkgreen</FGCOLOR>
<PARALLEL>yes</PARALLEL>
<LABEL>yes</LABEL>
<BUMP>yes</BUMP>
</ANCHORED_ARROW>
For a feature on the forward strand:

For a feature on the reverse strand:

This example shows an anchored arrow drawn orthogonal to the sequence map,
i.e. with <PARALLEL>no</PARALLEL>:
<ANCHORED_ARROW>
<HEIGHT>10</HEIGHT>
<FGCOLOR>darkgreen</FGCOLOR>
<PARALLEL>no</PARALLEL>
<LABEL>yes</LABEL>
<BUMP>yes</BUMP>
</ANCHORED_ARROW>
For a feature on the forward strand:

For a feature on the reverse strand:

BOX
A rectangular box covering the extent of the feature.
Attributes
| Attribute | Type | description |
|---|
| LINEWIDTH |
INT |
The width of the box outline (stroke). |
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The stroke (outline) color of the box. |
| BGCOLOR |
COLOR |
The fill color within the box. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
A green box with a red outline:
<BOX>
<FGCOLOR>red</FGCOLOR>
<BGCOLOR>chartreuse</BGCOLOR>
<BUMP>yes</BUMP>
<HEIGHT>10</HEIGHT>
<LABEL>yes</LABEL>
</BOX>

CROSS
A cross "+", commonly used for point mutations and other point-like features.
The width of the cross is not proportional by the size of the feature. Instead,
the cross is drawn at the centre of the feature and the width is always the
same as the height.
Attributes
| Attribute | Type | description |
|---|
| HEIGHT |
INT |
The absolute height (and width) of the glyph |
| FGCOLOR |
COLOR |
The color of the cross. |
| BGCOLOR |
COLOR |
The color of the background upon which the glyph is drawn. If omitted, the background is transparent. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Examples
A red cross on a cyan background:
<CROSS>
<FGCOLOR>red</FGCOLOR>
<BGCOLOR>cyan</BGCOLOR>
<BUMP>yes</BUMP>
<HEIGHT>10</HEIGHT>
<LABEL>yes</LABEL>
</CROSS>

As above, but for a feature whose extent is smaller than the cross:

As above, but with no <BGCOLOR> element.

DOT
Identical to the cross, with a circular dot drawn at the centre of the feature.
Attributes
| Attribute | Type | description |
|---|
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The color of the dot. |
| BGCOLOR |
COLOR |
The color of the background upon which the glyph is drawn. If omitted, the background is transparent. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
A red dot on an orange background:
<DOT>
<FGCOLOR>red</FGCOLOR>
<BGCOLOR>orange</BGCOLOR>
<BUMP>yes</BUMP>
<HEIGHT>10</HEIGHT>
<LABEL>yes</LABEL>
</DOT>

EX
Identical to the cross, with an "X" drawn at the centre of the feature.
Attributes
| Attribute | Type | description |
|---|
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The color of the "X". |
| BGCOLOR |
COLOR |
The color of the background upon which the glyph is drawn. If omitted, the background is transparent. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
"X" marks the centre of a feature whose extent is represented by the blue background:
<EX>
<FGCOLOR>red</FGCOLOR>
<BGCOLOR>lightblue</BGCOLOR>
<BUMP>yes</BUMP>
<HEIGHT>10</HEIGHT>
<LABEL>yes</LABEL>
</EX>

HIDDEN
A feature that is invisible, intended to support semantic zooming
schemes in which a feature is hidden at particular zooms. Hidden glyphs are
not rendered at all, but do affect the extent of a group where relevant.
Attributes: none.
LINE
A thin line, drawn parallel to the sequence object in the centre of a background
box. It is commonly used for parent features to indicate meaningful gaps
between child features, e.g. the introns between exons in a transcript.
Attributes
| Attribute | Type | description |
|---|
| STYLE |
ENUM (hat, solid, dashed) |
The line type. A type of "hat" draws an inverted V
(commonly used for introns). A type of "solid"
draws a horizontal solid line in the indicated color. A type of
"dashed" draws a dashed horizonal line in the indicated color. |
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. Note that this is the
height of the background, and not the thickness of the line.
|
| FGCOLOR |
COLOR |
The color of the line. |
| BGCOLOR |
COLOR |
The color of the background box. If omitted, it should be drawn with no color, i.e. transparent. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Examples
A simple solid line:
<LINE>
<FGCOLOR>sienna</FGCOLOR>
<STYLE>solid</STYLE>
<BUMP>yes</BUMP>
<HEIGHT>10</HEIGHT>
<LABEL>yes</LABEL>
</LINE>

In this image, the line represents a parent feature (e.g. a transcript) and
the boxes are parts (e.g. exons). Notice that the client is using a dashed
line to indicate there are other 'part' features beyond the edge of the
display. This is because the parent feature extends beyond the edge.

As above, but with <STYLE>hat</STYLE>:

As above, but with <STYLE>dashed</STYLE>:

SPAN
A line drawn across the extent of the feature and parallel to the reference
object, with orthogonal lines at each end.
Attributes
| Attribute | Type | description |
|---|
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The color of the lines. |
| BGCOLOR |
COLOR |
The color of the background upon which the lines are drawn. If omitted, the
background is transparent. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
<SPAN>
<FGCOLOR>darkgreen</FGCOLOR>
<BUMP>yes</BUMP>
<HEIGHT>10</HEIGHT>
<LABEL>yes</LABEL>
</SPAN>

TEXT
A static (pre-defined) ASCII string.
Attributes
| Attribute | Type | description |
|---|
| FONT |
FONT |
The font to use for the string. |
| FONTSIZE |
FONT |
The size (pt) of the font to use for the string. |
| STRING |
STRING |
The text to be rendered (required). |
| STYLE |
ENUM (bold, italic, underline) |
The font style to use for the string. Note that only one value is permitted per STYLE tag, but multiple STYLE tags may be present. |
| FGCOLOR |
COLOR |
The color of the text. |
| BGCOLOR |
COLOR |
The color of the background upon which the text is drawn. If omitted, the
background is transparent. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Examples
Purple text on a green background:
<TEXT>
<STRING>Some static text</STRING>
<FONT>courier</FONT>
<FONTSIZE>10</FONTSIZE>
<FGCOLOR>purple</FGCOLOR>
<BGCOLOR>green</BGCOLOR>
<LABEL>no</LABEL>
<BUMP>yes</BUMP>
</TEXT>

As above but with <LABEL>yes<LABEL>:

PRIMERS
Two inward-pointing arrows connected by a line (typically of a different
color). Used for showing primer pairs and a PCR product. The length of the
arrows is static - it is not related to the size of the feature.
Attributes
| Attribute | Type | description |
|---|
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The color of the arrows. |
| BGCOLOR |
COLOR |
The color of the connecting line. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
<PRIMERS>
<HEIGHT>10</HEIGHT>
<FGCOLOR>orange</FGCOLOR>
<BGCOLOR>limegreen</BGCOLOR>
<LABEL>yes</LABEL>
<BUMP>yes</BUMP>
</PRIMERS>

TOOMANY
Too many features than can be shown. Recommended for use in
consolidating sequence homology hits. The recommended visual
presentation is a set of overlapping boxes. The glyph is always drawn in the
center of its range, its height and width are not governed by the extent of
the feature but rather can be controlled by HEIGHT and LINEWIDTH respectively.
The background of the glyph covers the extent of the feature, but is
transparent unless a different colour is specified.
Attributes
| Attribute | Type | description |
|---|
| LINEWIDTH |
INT |
Width of the glyph |
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The stroke (outline) color. |
| BGCOLOR |
COLOR |
The fill color. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
<TOOMANY>
<HEIGHT>10</HEIGHT>
<FGCOLOR>purple</FGCOLOR>
<BGCOLOR>mediumorchid</BGCOLOR>
<LABEL>yes</LABEL>
<BUMP>yes</BUMP>
</TOOMANY>

TRIANGLE
A triangle. Commonly used for point mutations and other point-like
features. The triangle is always drawn in the center of its range,
its width and height are not governed by the extent of the feature but rather
can be controlled by HEIGHT and LINEWIDTH respectively.The orientation of the
triangle is determined by its DIRECTION, not the orientation (strand) of the
features. The background of
the glyph covers the extent of the feature, but is transparent unless a
colour is specified.
Attributes
| Attribute | Type | description |
|---|
| DIRECTION |
ENUM |
Indicates the direction of the triangle's apex, assuming the segment is represented in the west-east plane. One of "N", "E", "S", and "W". |
| LINEWIDTH |
INT |
The absolute width of the glyph, in pixels. |
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| FGCOLOR |
COLOR |
The colour of the triangle. |
| BGCOLOR |
COLOR |
The colour of the background behind the triangle, covering the extent of
the feature. If this element is omitted, the background is transparent. |
| LABEL |
BOOL |
Whether the glyph should be labeled with its name, as dictated
by the <FEATURE> label attribute in the DASGFF document. |
| BUMP |
BOOL |
Whether the glyph should "bump" intersecting glyphs (e.g. onto the next line) to prevent them from overlapping. Note: This attribute only applies to feature glyphs and not to groups. Groups of features will always bump. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Examples
The below examples show a series of triangles with directions of north, south,
east and west respectively. All triangles are 5 pixels wide and 10 pixels high.
<TRIANGLE>
<DIRECTION>N</DIRECTION>
<FGCOLOR>green</FGCOLOR>
<BGCOLOR>orange</BGCOLOR>
<HEIGHT>10</HEIGHT>
<LINEWIDTH>5</LINEWIDTH>
<LABEL>yes</LABEL>
<BUMP>yes</BUMP>
</TRIANGLE>

As above but with no BGCOLOR element and therefore no background covering the
extent of the feature.
<TRIANGLE>
<DIRECTION>N</DIRECTION>
<FGCOLOR>green</FGCOLOR>
<HEIGHT>10</HEIGHT>
<LINEWIDTH>5</LINEWIDTH>
<LABEL>yes</LABEL>
<BUMP>yes</BUMP>
</TRIANGLE>

GRADIENT
A colour gradient. This is an orthogonal oblong covering the extent of the
feature, coloured according to the value of the feature's score tag.
The exact colour is assigned proportionally by the client, according to a range
of possible scores and colours as supplied by the server. If no minimum or
maximum values are supplied, the client will assign them dynamically from all the
available features.
Attributes
| Attribute | Type | description |
|---|
| HEIGHT |
INT |
The absolute height of the glyph, in pixels. |
| COLOR1 |
COLOR |
The colour of the glyphs for the lowest-scoring features. |
| COLOR2 |
COLOR |
The colour of the glyphs for the medium-scoring features. |
| COLOR3 |
COLOR |
The colour of the glyphs for the highest-scoring features. |
| MIN |
INT or FLOAT |
The minimum score cutoff. Scores below this value will be rounded up. |
| MAX |
INT or FLOAT |
The maximum score cutoff. Scores above this value will be rounded down. |
| STEPS |
INT |
The number of steps, or grades of colour, to use across the whole gradient.
Note that the number of steps can be greater than the number of
explicitly defined colours. It is usually used to restrict the number
of possible shades. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
The following example defines a histogram where features are drawn in a colour
determined according to where their scores lie in a -50 to 50 range. Scores
outside the range are rounded appropriately. The
colour gradient moves from red (lowest scores) through the spectrum via
yellow to blue (highest scores) with a total of 50 possible colours (steps).
Thus glyphs may be different shades of red, orange, yellow, green and blue.
The histogram as a whole has a total height of 40 pixels.
<GRADIENT>
<MIN>-50</MIN>
<MAX>50</MAX>
<HEIGHT>40</HEIGHT>
<STEPS>50</STEPS>
<COLOR1>red</COLOR1>
<COLOR2>yellow</COLOR2>
<COLOR3>blue</COLOR3>
</GRADIENT>

Special notes
- Colour gradient glyphs always bump.
- Colour gradient glyphs never have labels (though their parents may).
HISTOGRAM
A histogram plot. This glyph has all the properties of a colour gradient, and
in addition the height is proportional to the value of the feature's score tag.
The exact height is determined in the same way as the colour, and can be a
negative value if the score is less than zero. A single-colour
histogram can be easily specified by supplying only one colour.
Attributes
| Attribute | Type | description |
|---|
| HEIGHT |
INT |
The absolute height of the histogram, in pixels. |
| COLOR1 |
COLOR |
The colour of the glyphs for the lowest-scoring features. |
| COLOR2 |
COLOR |
The colour of the glyphs for the medium-scoring features. |
| COLOR3 |
COLOR |
The colour of the glyphs for the highest-scoring features. |
| MIN |
INT or FLOAT |
The minimum score cutoff. Scores below this value will be rounded up. |
| MAX |
INT or FLOAT |
The maximum score cutoff. Scores above this value will be rounded down. |
| STEPS |
INT |
The number of steps, or grades of colour, to use across the whole gradient.
Note that the number of steps can be greater than the number of
explicitly defined colours. It is usually used to restrict the number
of possible shades. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
The following example defines a histogram where features are drawn in a colour
determined according to where their scores lie in a -50 to 50 range. Scores
outside the range are rounded appropriately. The
colour gradient moves from red (lowest scores) through the spectrum via
yellow to blue (highest scores) with a total of 50 possible colours (steps).
Thus glyphs may be different shades of red, orange, yellow, green and blue.
At the same time, the height of each glyph is also determined by the score.
Because values < 0 are possible in this example, the y axis is split
between positive and negative. The histogram as a whole has a total height of
20 pixels.
<HISTOGRAM>
<MIN>-50</MIN>
<MAX>50</MAX>
<HEIGHT>20</HEIGHT>
<STEPS>50</STEPS>
<COLOR1>red</COLOR1>
<COLOR2>yellow</COLOR2>
<COLOR3>blue</COLOR3>
</HISTOGRAM>

Special notes
- Histogram glyphs always bump.
- Histogram glyphs never have labels (though their parents may).
LINEPLOT
A point-to-point line plot. This glyph has all the properties of a colour
gradient, except features are represented as joined points on a graph rather
than boxes. A single-colour lineplot can be easily specified by supplying only
one colour.
Attributes
| Attribute | Type | description |
|---|
| HEIGHT |
INT |
The absolute height of the plot, in pixels. |
| COLOR1 |
COLOR |
The colour of the glyphs for the lowest-scoring features. |
| COLOR2 |
COLOR |
The colour of the glyphs for the medium-scoring features. |
| COLOR3 |
COLOR |
The colour of the glyphs for the highest-scoring features. |
| MIN |
INT or FLOAT |
The minimum score cutoff. Scores below this value will be rounded up. |
| MAX |
INT or FLOAT |
The maximum score cutoff. Scores above this value will be rounded down. |
| STEPS |
INT |
The number of steps, or grades of colour, to use across the whole gradient.
Note that the number of steps can be greater than the number of
explicitly defined colours. It is usually used to restrict the number
of possible shades. |
| ZINDEX |
INT |
The rendering order of the glyph in the Z dimension, useful for overlapping glyphs that do not bump. Glyphs with higher ZINDEX values are drawn "on top" (i.e. closer to the user). |
Example
<LINEPLOT>
<MIN>-50</MIN>
<MAX>50</MAX>
<HEIGHT>50</HEIGHT>
<STEPS>50</STEPS>
<COLOR1>red</COLOR1>
<COLOR2>yellow</COLOR2>
<COLOR3>blue</COLOR3>
</LINEPLOT>
Special notes
- Line plot glyphs always bump.
- Line plot glyphs never have labels (though their parents may).
Below are a list of changes to the specification. Developers wishing to update
their software are encouraged to review their application in the context of
the entire specification document, as clarifications of previously vauge
or misinterpreted features are not listed here.
Version 1.6
This version mainly introduces concepts and extensions that are already in use
but yet to be incorporated into the specification.
- Nonpositional annotations are now supported.
- Introduced the concept of "coordinate systems" and the DAS Registry.
- Relaxed the constraints on data source names.
- Clarified the use of HTTP and DAS status codes.
- Clarified content encodings and compression.
- Added requirement for clients to include specific request headers.
- All command responses are now described by RELAX NG schemas.
- New commands: sources and structure. These commands are taken from DAS 1.53E, with some changes.
- Deprecated commands: dsn, dna and link.
- Entry points command gained "paging", and other minor changes.
- Features command gained hierarchical referencing, in favour of groups.
- Segment query parameters no longer require start and end positions.
- Several elements in the features command response are now optional.
- Features command gained "maxbins".
- Unified the format of segment XML across all commands.
- Added support for using ontologies in annotations.
- Stylesheets now support histograms, colour gradients and line plots, more colours, and are better characterised. The "toomany" glyph is deprecated.
- Clarified the content of attributes across several commands, such as segment versions.
Version 1.51
- The description of the entry_points document was out of synch
with the DTD. Also there seems to have been some semantic drift between
Dazzle, the UCSC server, and LDAS with regards to the attributes of the
<SEGMENT> tag. This has now been made explicit, and the DTD relaxed
to allow all styles.
Version 1.5
- Added capabilities header.
- Added exception handling for invalid sequence IDs.
- Added feature_id request.
- Corrected syntax errors in stylesheet example.
Version 1.01
- Split assembly functionality into "component" and "supercomponent".
- Removed redundant descriptions of glyph attributes.
Version 1.0
- Removed deprecated resolve command.
- Removed deprecated entry_points ref argument.
- Added superparts attribute to DASGFF <FEATURE> tag.
- New discussion of how to move upwards in an assembly.
- Reorganized specification to put responses close to requests.
- Added a stylesheet example document.
- Normalized the names of glyph COLOR and FILLCOLOR attributes to
FGCOLOR and BGCOLOR.
- Added the LABEL attribute to all glyphs.
- Added the STYLE attribute to the LINE glyph.
- Added the ability to assign a glyph to a group.
- Added HIDDEN glyph.
Version 0.999
- Added LINK, NOTE, and TARGET to FEATURE
- Added section entitled "Fetching Sequence Assemblies"
Version 0.998
- Deprecated regular expression matching for types and categories.
- Allow multiple TYPE arguments for logical OR filtering.
- Made FEATURE optional within features return document.
- Made TYPE optional within types return document.
Version 0.996
- Added subparts tag to features and entry_points.
- Removed the requirement that the server return features that
do not overlap with the requested segment.
- Added support for multiple segments/sequences in types document.
Version 0.995
- Added support for multiple segments/sequences in returned documents.
- Added support for assembly components.
Version 0.99
- Allow query parameters to be POSTed to the DAS URL.
- Added compatibility warning about SOAP conversion.
- Use Version 8 regular expressions rather than GNU's, giving
compatibility with both Perl regex and GNU regex.
- Made the id attribute of the <TYPE> tag required.
- Changed the WIDTH glyph attribute to HEIGHT throughout.
Andrew M. Jenkinson, andy.jenkinson@ebi.ac.uk
European Bioinformatics Insitute
Last modified: Thu Nov 18 12:26:00 GMT 2010
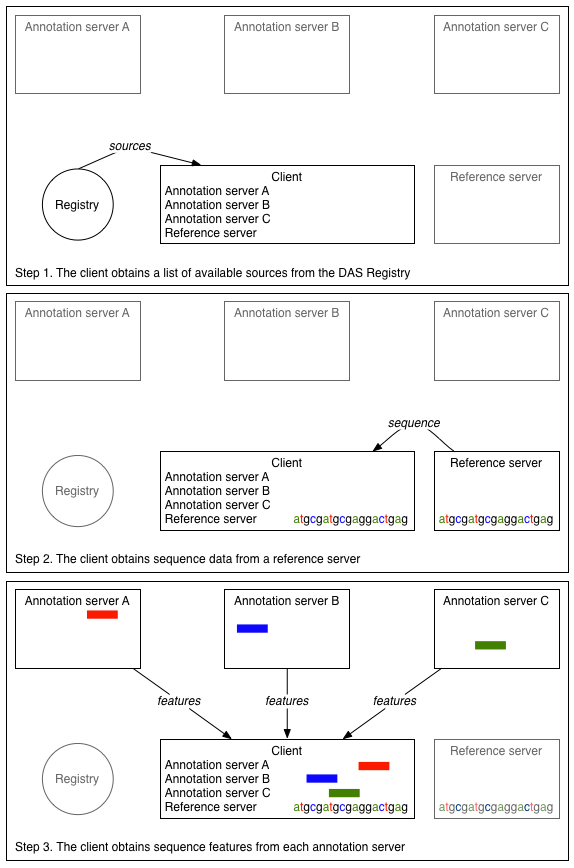{kind=link}